58-jähriger Patient mit neu diagnostiziertem Diabetes und Gewichtsverlust
Anamnese
Vorstellung mit einer im auswärtigen Computertomogramm gesehenen Pankreaskopf-Raumforderung mit Erweiterung des Ductus wirsungianus.
Initial war der Patient ärztlich aufgrund einer Gewichtsabnahme nach Erstdiagnose eines Diabetes mellitus vorstellig gewesen.
Labor
| Parameter | Werte | Normbereich |
|---|---|---|
| Natrium | 140 | 135 – 145 mmol/l |
| Kalium | 4.13 | 3,5 – 4,8 mmol/l |
| Calcium | 2.03 | 2,1 – 2,65 mmol/l |
| Kreatinin | 0.88 | 0,1 – 1,3 mg/dl |
| Glucose | 119 | 65 – 110 mg/dl |
| GOT/AST | 20 | – 35 U/l |
| GPT/ALT | 24 | – 35 U/l |
| AP | 49 | 40 – 130 U/l |
| GGT | 36 | – 40 U/l |
| Dir.Bili. | 0.13 | – 0,3 mg/dl |
| P-Amylase | 35 | 8 – 53 U/l |
| Lipase | 64 | – 51 U/l |
| Gesamteiweiß | 67.8 | 60 – 80 g/l |
| Albumin | 41,8 | 30 – 50 g/l |
| CRP | <2.0 | – 5 mg/l |
| Quick | 109.6 | 70 – 125 % |
| INR | 0.940 | – 1.2 |
| aPTT | 22,9 | – 35 s |
| CA 19-9 | 24,9 | – 38,0 U/ml |
| CEA | 1,9 | – 2,5 µg/l |
Bildgebung
Sono- und Endosonographie: Darstellung einer Raumforderung im Pankreaskopf/-korpus von ca. 1,5 cm.
CT-Abdomen:
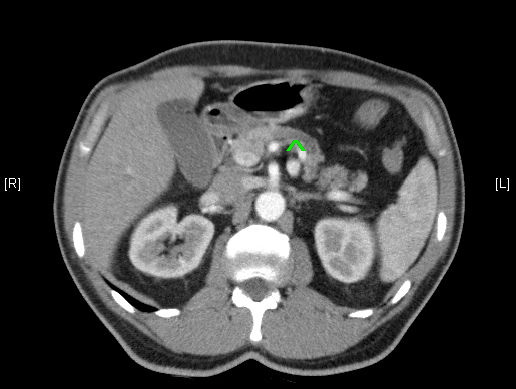
Abb. 379: CT-Abdomen, arterielle Phase nach KM-Gabe; Im Pankreaskorpus ist eine max. 1,8 cm große hypodense Raumforderung erkennbar, keine KM Aufnahme, damit hochgradig malignomsuspekt.
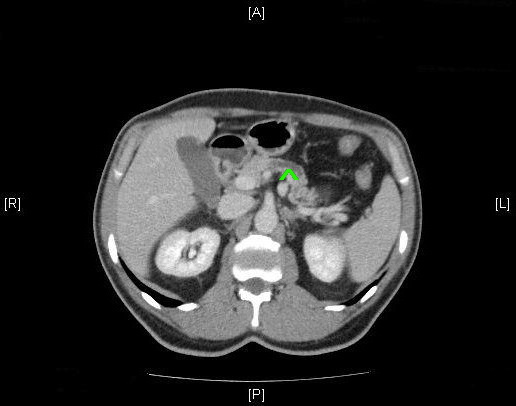
Abb. 380: CT-Abdomen (nativ); Erkennbar ist eine hypodense Raumforderung von ca. 1,8 cm im Pankreaskorpus.
Die Umfelddiagnostik war unauffällig.
Procedere
Aufgrund der o. g. Befunde stellten wurde die Indikation zur Exploration mit dem Ziel einer Resektion des Tumors. Die Bauchraumerkundung zeigte keinen Hinweis für Lebermetastasen oder eine Peritonealkarzinose, es folgte eine pyloruserhaltende OP nach Wipple. Im Schnellschnitt wurde der Verdacht auf ein neuroendokrines Karzinom geäußert.
Makroskopie
Ein erweitertes Pylorus-erhaltendes Whipplepräparat mit 30 cm Dünndarm, 12 × 7 x max. 4 cm messenden Pankreaskopf-/corpus, einem 5 cm langen Ductus choledochus, einer 8 cm langen, dünnwandigen, Gallenblase mit 4 cm langem Ductus zysticus. Im Pankreascorpus, 1 cm vom Schnittrand entfernt ein im Maximum 1,8 cm großer, grau-gelber, scharf begrenzter durchmessender Tumor.
Virtuelle Mikroskopie
Mikroskopie
Solide, trabekulär und kleinherdig pseudoglanduläre Formationen wenig pleomorpher mittelgroßer Tumorzellen, die in eine bindegewebige Grundsubstanz eingebettet sind und abschnittsweise hellzelliges Zytoplasma aufweisen. In 10 hochauflösenden Gesichtfeldern finden sich bis zu 4 Kernteilungsfiguren.
Immunhistochemie
Immunhistochemisch exprimieren die Tumorzellen Synaptophysin und Chromogranin A, fokal Glukagon und einzelzellulär Somatostatin. Negativer Reaktionsausfall gegenüber Gastrin, Insulin, pankreatisches Polypeptid und Serotonin. Die Proliferationsaktivität (Ki-67) liegt bei 5%.
Diagnose
Gut differenziertes endokrines Pankreaskarzinom
Postoperativer Verlauf
Der postoperative Verlauf gestaltete sich komplikationslos. Es bestanden keine Verdauungsbeschwerden; der Stuhlgang war regelrecht.
Laborchemisch imponierten postoperativ zunächst erhöhte Infektwerte, die im Verlauf jedoch regredient und bei Entlassung schließlich im Normbereich lagen. Die Blutzuckerkonzentration war postoperativ von Beginn an nahezu im Normbereich.
Eine adjuvante Therapie war aufgrund der G2- und R0-Resektion bei neuroendokrinem Tumor nicht indiziert.
Die erste postoperative Nachsorge erfolgte ein viertel Jahr spater. Hier gab der Patient noch starken Meteorismus und teilweise starker Erschöpfung an, bei sonst gutem Wohlbefinden. In der letzten Zeit hat der Patient 3 kg an Gewicht zugenommen.
Die durchgeführte Diagnostik ergab im CT-Thorax keinen Nachweis suspekter Lungenrundherde. Das durchgeführte Abdomen-CT ergab eine regelrechte Darstellung des Restpankreasparenchyms ohne Anhalt für ein Lokalrezidiv. Keine suspekten Leberläsionen.
Die Laborkontrolle ergab keine Erhöhung der Tumormarker (CEA 2,2 und CA19-9 10,0).
Kommentar
Endokrine maligne Pankreastumoren sind im Vergleich zur Häufigkeit exokriner Malignome des Pankreas insgesamt selten (etwa 1% aller Pankreasmalignome).
Diese Tumoren treten häufiger sporadisch als im Rahmen eines genetischen Syndroms auf. Als genetische Prädisposition gelten in diesem Kontext das MEN 1-Syndrom und die von Hippel-Lindau Erkrankung. Vor allem bei genetischen Syndromen können endokrine Pankreastumoren schon im Kindesalter auftreten, häufiger sind sie jedoch im Erwachsenenalter.
Unterteilt werden diese Tumoren in funktionell aktive (selten) und inaktive (häufig) Läsionen, die Unterscheidung erfolgt aufgrund der klinisch detektierbaren Hormonaktivität.
Nach WHO unterscheidet man bei den endokrinen Pankreastumoren Tumoren mit voraussichtlich benignem Verhalten (Größe < 2 cm, geringe Proliferationsaktivität) von malignen Tumoren. Selten sind schlecht differenzierte endokrine Karzinome des Pankreas mit mehr als 10 Mitosen in 10 HPF.
Kasuistiken

Bilder
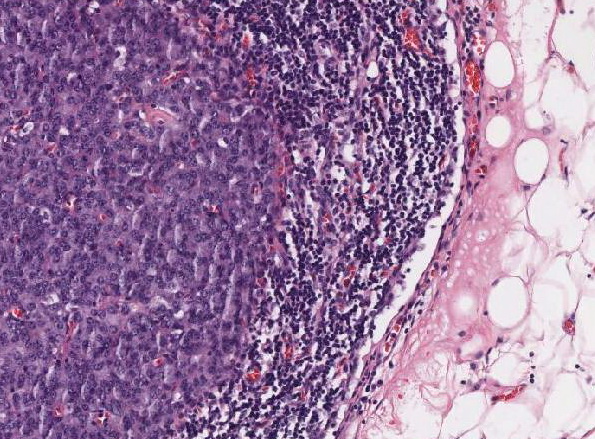
Abb. 439: Lymphknotenmetastase mit solider Tumordifferenzierung
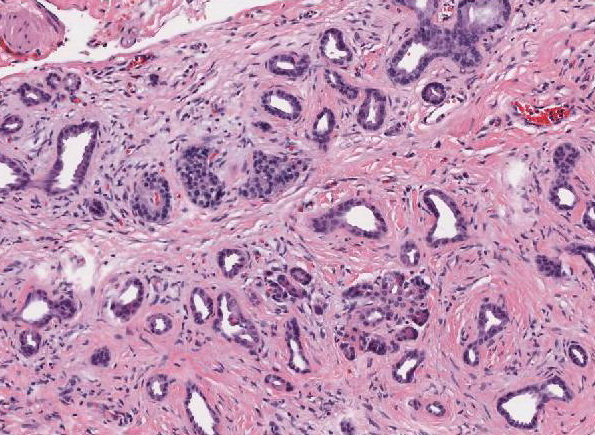
Abb. 440: Pseudoglanduläre Tumorformationen und spärliches, atrophes exokrines Pankreasparenchym
Pankreas - Lehrtexte
- Chronische Pankreatitis
- Duktales Adenokarzinom des Pankreas
- Insulinom
- Intraduktal papillär muzinöse Neoplasie (IPMN)
- Intraduktal papillär muzinöse Neoplasie (IPMN) Steckbrief
- M. Whipple
- Neuroendokrine Tumoren des Pankreas
Pankreas - weitere Kasuistiken
Pankreas - Literatur
Organpathologie-Atlas