57-jähriger Patient mit Raumforderung in der linken Kniekehle
Anamnese
Zunehmende indolente Schwellung in der linken Kniekehle. Ein Trauma ist nicht erinnerlich. Bei Bewegung und Belastung bestehen Schmerzen im Bereich der linken Kniekehle, deren Intensität in den letzen Wochen zugenommen hatte. Sonst gutes Allgemeinbefinden. Kein Nachtschweiß, kein Gewichtsverlust, keine Infekt-, Blutungs oder Thrombosezeichen.
Bildgebende Diagnostik
Im MRT (T2-gewichtet) eine 6×4×3,5cm große, solide extraartikuläre popliteale Raumforderung ohne Gefäß- oder Nervenbezug:
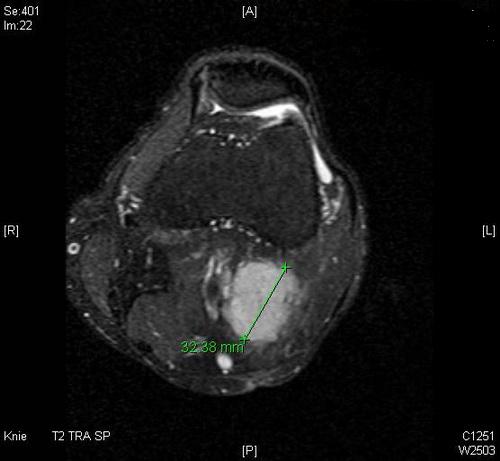
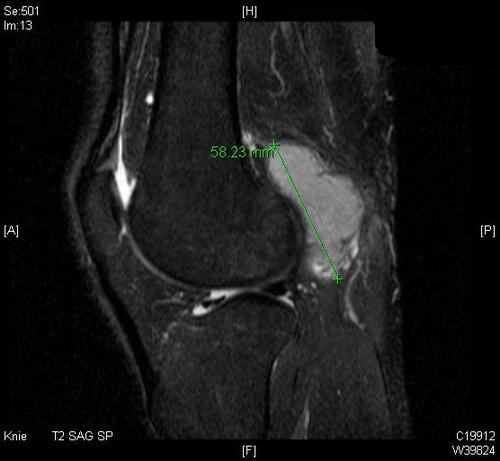
CT: Die Thorax und die Abdomenaufnahme ergaben im Staging kein Hinweis auf eine Metastasierung.
Procedere
Diagnostische inzisionale Biopsie. Beginn einer neoadjuvanten Chemotherapie (2-4 Zyklen) nach dem EIA-Schema (Adriamycin, Etoposid und Ifosfamid) gefolgt von einer Tumorresektion mit einer intraoperativen Bestrahlung und einer adjuvanten Chemotherapie und Bestrahlung.
Makroskopie
9,5 × 9 × 4,5cm messendes aus Fettgewebe und spärlich Skelettmuskelgewebe bestehendes Weichgewebsresektat in Zusammenhang mit einer 5,5 × 2cm messenden Hautspindel mit zentraler, 3 cm langer, verheilter chirurgischer Inzisionsnarbe. Auf den Schnittflächen im Fettgewebe eine 7 × 4 × 3,5cm messender, vergleichsweise zirkumskripter, rot-braun gescheckter, relativ weicher Tumor, nicht randbildend. Im Fettgewebe miterfasst ein 0,7 cm großer Lymphknoten.
Virtuelle Mikroskopie
HE-Färbung
Diagnose
Mittelgradig differenziertes biphasisches Synovialsarkom. Der Lymphknoten ist tumorfrei.
Mikroskopie
Innerhalb des subkutanen Fettgewebes teilweise mit Bezug zur tiefen Faszie ein knotig gestalteter flächenhaft vitaler biphasischer maligner Tumor. Erkennbar sind spindelig ausgezogene Tumorzellen mit ovalären bis länglichen bipolaren Kernen und einem zart ausgezogenen Zytoplasmasaum. Das Tumorgewebe überwiegend zellreich mit kleinen pseudozystischen Aufweitungen. In der Tumorpopulation gelegentlich Mitosen (weniger als 10 Mitosen pro 10 HPF) Keine Tumornekrosen.
Tumorstadium
TNM (6. Aufl.): ypT2b, ypN0 (0/1)
R-Klassifikation: R0
Klinischer Verlauf
Es folgte eine postoperative Radiotherapie der Tumorregion mit einer Gesamtdosis von 41,4 Gy sowie 4 Zyklen adjuvanter EIA-Chemotherapie gemäß dem Neo-WTS-Protokoll.
Die Tumornachsorge ein Jahr postoperativ zeigt weder klinisch noch bildgebend ein Hinweis für ein Rezidiv der Erkrankung bzw. für eine pulmonale Metastasierung.
Eine weitere Verlaufskontrolle einschließlich MRT des Lokalbefundes sowie ein CT des Thorax erfolgte 5 Monate später und ergab ebenfalls kein Hinweis auf ein Rezidiv.
Diagnostische Kriterien
Im biphasischen Synovialsarkom finden sich häufig Kombinationen aus epithelialen, glandulären seltener auch sqamoiden sowie spindelzelligen fibroblastenartigen Komponenten. Im monophasich fibrösen Synovialsarkom findet sich dagegen häufig ein fischgrätenartiges Wachstummuster oder eine Kernpalisadierung wie bei neurogenen Tumoren sowie eine myxoide Tumorkomponente. Hyalinisierungen, Kalzifizierungen oder auch eine metaplastische Ossifikation sind möglich.
Gering differenzierte Synovialsarkome können aus morphologisch undifferinzierten Tumorzellen bestehen, die konventionell-morphologisch nicht immer von anderen Sarkomtypen (z.B.: Ewing Sarkom, oder Rhabdomyosarkom) oder gar von malignen Lymphomen oder kleinzelligen Karzinomen abgegrenzt werden können und erst unter Zuhilfenahme immunhistochemischer und molekularpathologischer Zusatzuntersuchungen digagnostisch gesichert werden können.
Klinische Korrelation
Synovialsarkome sind epithelial/mesenchymal differenzierte Tumoren und gehören mit etwas 7-10% zu den vierthäufigsten Weichteilsarkomen nach dem MFH, Liposarkom und dem Rhabdomyosarkom. Sie können in jedem Lebensalter auftreten, Kinder miteingeschlosssen.
Histologisch werden biphasische, monophasisch spindelzellige (fibröse) und gering differenzierte Synovialsarkome unterschieden. Monophasisch epitheliale Synovialsarkome stellen eine Rarität dar und dürfen nicht mit epthelial differentzierten Malignome (Karzinomen) verwechselt werden.
In deutlich über 95% der Synovialsarkome ist die für diesen Sarkomtyp charakteristische balancierte Transloktion t(X;18)(p11.2;q11.2) nachweisbar, die zu einer Fusion des SS18 (SYT) Gens auf Chromosom Xp11.2 mit einem der SSX Gene auf Chromosom 18q11.2 führt. (SSX1 oder SSX2, sehr selten auch SSX4)
Lokalrezidive entstehen nur selten und dann zumeist nach inkompletter mirkroskopischer (R1) oder makroskopischer (R2) Resektion. Fernmetastasen insbesondere in den Lungen und den regionären LK treten im Verlauf der Erkrankung bei bis zu 50% der Betroffenen auf. Die Prognose dieser Patienten in Bezug auf das 5-Jahres Überleben beträgt weniger als 10%.
Literatur
Radiologie beim Synovialsarkom
Murphey MD, Gibson MS, Jennings BT, et al: From the archives of the AFIP: Imaging of synovial sarcoma with radiologic-pathologic correlation. Radiographics. 2006 Sep-Oct;26(5):1543-65Synovialsarkom in der klinischen Praxis
Przkora R, Vogel P, Knüchel R, Jauch KW, Bolder U. Das Synovialsarkom in der klinischen Praxis--eine Zusammenstellung ausgesuchter Fälle. Zentralbl Chir. 2003 Mar;128(3):239-4Diagnosis and management of synovial sarcoma
Eilber FC, Dry SM. Diagnosis and management of synovial sarcoma. J Surg Oncol. 2008 97: 314-20.
Kasuistiken

Bilder
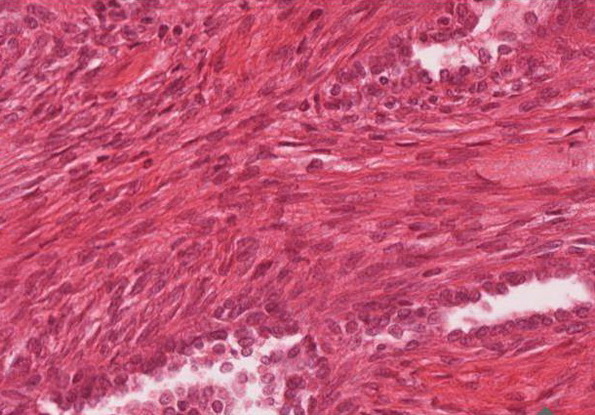
Abb. 207:
Weichgewebe - Lehrtexte
Weichgewebe - weitere Kasuistiken
Weichgewebe - Literatur
Organpathologie-Atlas