Sarkoidose
Definition
Die Sarkoidose ist eine Multisystemerkrankung unklarer Ursache.2 Die Erkrankung ist gekennzeichnet durch die Bildung von nichtverkäsenden,4 epitheloidzelligen6 Granulomen in den betroffenen Organen.2 Die Lunge und das lymphatische System sind dabei am häufigsten betroffen.2 Zu einem Befall der Haut, Augen, peripheren Lymphknoten und Leber kann es in ca. 10-25% der Fälle kommen.2 Da die Sarkoidose ein sehr heterogenes Krankheitsbild ist, und neben oben genannten Organen noch andere Manifestationsorte hat,6 wird sie sehr gerne als Cameleon bezeichnet.Epidemiologie
Die Sarkoidose zählt trotz niedriger Prävalenz zu den häufigsten interstitiellen Lungenerkrankungen.6 Sie betrifft vor allem junge Patienten vor dem 50. Lebensjahr mit einem Erkrankungsgipfel im Alter von 20-39 Jahren.1 Die Inszidenz ist weltweit unterschiedlich und variiert je nach Geschlecht, Alter, Rasse und geografischer Herkunft. Sie wird weltweit auf 16,5/100000 bei Männern und 19/100000 bei Frauen geschätzt2 und ist mit 5-40 Fälle pro 100000 Einwohnern in Nordeuropäischen Länder am höchsten.1 Unter 15 Jahren ist die Sarkoidose mit 1/100000 Fällen selten, unter 4 Jahren (Early onset sarcoidosis, EOS) sogar verschwindend gering. (0,06/100000)2 Die Sarkoidose tritt für gewöhnlich sporadisch auf, eine familiäre Häufung wurde in 1,7-17% der Fälle beobachtet.2 Weiterhin konnte belegt werden, dass es für die Sarkoidose eine genetische Prädisposition gibt.6 Eine Genmutation im Eiweiß BTNL2 auf Chromosom 6 steht mit einem erhöhten Risiko an Sarkoidose zu erkranken in Verbindung.9 Mutationen des CART 15-Gens auf Chromosom 16p12-q21prädisponieren für eine familiäre (Blau-Syndrom) oder spontanen Form der frühen Sarkoidose (EOS).9
Pathogenese
Die genaue Ursache der Sarkoidose ist nach wie vor nicht bekannt.1 Ein Merkmal der Erkrankung ist die Anwesenheit von CD4 positiven Zellen die mit antigenpräsentierenden Zellen interagieren und so die Bildung von Granulomen fördern.1 Aktivierte CD4 positive Zellen differenzieren im weiteren Verlauf zu TH1- Zellen.1 Dabei werden insbesondere die zellulären Botenstoffe TNF α, Interferon γ und Interleukin 2 von den Entzündungszellen deutlich vermehrt produziert.1 Die natürlichen Killerzellen sind im peripheren Blut hingegen vermindert, ihre Rolle im Rahmen der Sarkoidose ist noch ungeklärt.1
Neue Hinweise zeigen, dass Sarkoidosepatienten auf an sich harmlose Erreger, wie z. B. ubiquitär vorkommende atypische Mykobakterien und Propionibakterien, mit einer überschießenden Entzündungsantwort reagieren, so dass diese möglicherweise eine Rolle in der Pathogenese spielen könnten.6
Makroskopie
- Lymphknoten: Charakteristisch sind stark vergrößerte cervikale-, hiliäre-, und mediastinale Lymphknoten.7
- Lunge: Makroskopisch kommen die Graunulome in den Lungen häufig in Gruppen liegend zur Darstellung.8 Sie erscheinen als 2-3mm große grau-weiße Knötchen, nicht selten als 1-2 cm große Konglomeratherde, und finden sich bevorzugt peribronchial, paraseptal und pleural.8 Oft sind sie wie Glieder einer Kette, den Lymphwegen folgend, angeordnet.8 Selten können in sehr großen Konglomeratherden Kavernisierungen entstehen.8 Im Endstadium der Erkrankung zeigt die Lunge das Bild einer Fibrose.7
Mikroskopie
Granulome sind kompakt-organisierte histologische Strukturen.1 Zentral enthalten sie Ansammlungen von Makrophagen und Epitheloidzellen (sie können zu mehrkernigen Riesenzellen verschmelzen) die von einem Lymphozytensaum umgeben werden.1 Im weiteren Verlauf kann es in der Granulomperipherie zur Fibrosierung kommen, von denen aus Kollagenfaserbündel in das Innere ziehen und unter Hyalinisierung zunehmend breiter werden.8 Durch Konfluenz der Knötchen können sich größere Schwielenfelder bilden.8 Die Fibrosierung schreitet gewöhnlich hilifugal fort, mit Folge einer narbigen perihilären Schrumpfung des Lungengewebes einschließlich der hilären Lymphknoten.8 In der Peripherie entsteht so ein wabiger Umbau der Lunge mit linsen- bis erbsengroßen, durch derbes Bindegewebe begrenzten, glattwandigen Hohlräumen.8 Eine Lungenfibrose mit restriktiver Ventilationsstörung ist die Folge.8
Bestandteile eines Granuloms
- Epitheloidzellen: Sie sind ca. 30μm groß und enthalten einen langgestreckten hellen Zellkern.7 In den Epitheloidzellen ist Angiotensin-converting-enzyme (ACE) nachweisbar.8 Elektronenmikroskopisch ist das Zytoplasma der Zellen sehr organellenreich.8
- T-Lymphozyten: Sie sind in der Granulomperipherie reichlich enthalten.7
- Riesenzellen: Sie entstehen aus der Fusion mononukleärer Makrophagen und entsprechen zunächst einer Fremdkörperriesenzelle, die sich im Verlauf in eine Riesenzelle vom Langhans-Typ umwandelt.7
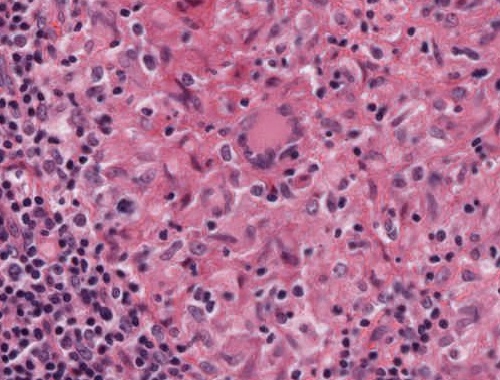
Abb. 467: Epitheloidzellgranulom (Detail) mit Riesenzelle vom Langhans-Typ
Typisch aber nicht pathognomonisch für Riesenzellen sind verschiedene Zytoplasmaeinschlüsse.7
- Schaumann-Körperchen: Hierbei handelt es sich um konzentrisch geschichtete bis 200μm große Ablagerungen, die sich aus einem kalziumoxalatimprägnierten Mukopolysaccaridkern zusammensetzen.7 Doppelbrechende Kristalle finden sich in 70% dieser Zellen.7 Bei 90% der Sarkoidosen sind Riesenzellen mit Schaumann-Körperchen nachweisbar.7
- Riesenzellen mit Asteroidkörperchen: Sie sind bis zu 30μm groß und enthalten sternförmige Einschlüsse.7 Asteroidbodies scheinen aus Mikrotubuli, Mikrofilamenten und Zentriolen zu entstehen.8 Die Zellen werden nur bei 10% der Sarkoidosen nachgewiesen.7 Sie kommen auch bei Lepra, Mykosen und Fremdkörpergranulomen vor.7
Die Granulome bei Sarkoidose lassen sich histologisch nicht eindeutig von Granulomen anderer Genese unterscheiden.1 Als differentialdiagnostische Abgrenzung gegenüber der Tuberkulose dient das Fehlen von Nekrosen bei der Sarkoidose.7 Dennoch kann es bei jedem dritten Patienten zur Ausbildung kleiner verkäsender Koagulationsnekrosen vor allem in größeren Granulomen kommen.7 Aus diesem Grund sind weitere histologische Aufarbeitungen des Gewebes nötig, so z.B.: Ausschluss säurefester Stäbchen oder Pilze.1
Klinik
Man unterscheidet eine akute Verlaufsform von einem nicht akuten, schleichenden Verlauf.6 Die Sarkoidose verläuft in vielen Fällen asymptomatisch und wird oft bei Röntgen-Thorax Aufnahmen im Rahmen der Routinediagnostik entdeckt.1 Typisch ist die Diskrepanz zwischen einem relativ guten subjektiven Befinden und ausgeprägten objektiven Befunden. (Röntgen Thorax)9
Das Löfgren-Syndrom ist die akute Manifestation der Sarkoidose und beinhaltet eine Sprunggelenksarthritis (häufiger bei Männern), ein Erythema nodosum (häufiger bei Frauen) und die bihiläre Lymphadenopathie.1,2 Zum Löfgren Syndrom kommt es in 9-34% der Fälle.1
Weitere häufige unspezifische Symptome umfassen Müdigkeit, Nachtschweiß, Gewichtsverlust, Fieber und Abgeschlagenheit.1,6 Betroffene Patienten haben häufig einen extrapulmonalen Befall.6
Organmanifestation
- Pulmonale Manifestation: Sie ist am häufigsten und umfasst Dyspnoe und trockener Husten.1,2
- Periphere Lymphadenopathie: Palpale Lymphknoten sind von fester Konsistenz und meist unterschiedlicher Größe.2 Ihre Häufigkeit variiert zwischen den einzelnen Studien.2 Sie finden sich am Hals, in den Axilla, der Leiste4 und im Abdomen.2
- Kutane Manifestationen: Sie treten bei ca. 25-35% der Patienten auf, werden häufig missinterpretiert und können als Maculae, Papeln und Plaques, einzeln oder in Gruppen in Erscheinung treten.1 Häufige Lokalisationen umfassen den Hals, den oberen Rücken, die Extremitäten und den Stamm.1 Bei Lupus pernio finden sich flächenhafte livide Infilrationen,9 die meist die Nase, Wangen, Lippen und Ohren betreffen und charakteristisch für die Sarkoidose sind1 Lupus pernio ist häufiger bei Frauen als bei Männern.1 Zu einem Erythema nodosum kommt bei bis zu 10% der Patienten.1 Dabei handelt es sich um subcutane rotbläuliche Knoten an den Streckseiten der Unterschenkel, die sehr druckschmerzhaft sind9 und gewöhnlich über einen Zeitraum von drei Wochen bestehen.1 Als weitere Hautmanifestation kann eine Narbensarkoidose vorliegen.9 Gelbbräunliche Plaques im Bereich bestehender Narben kennzeichnen diese.9
- Leber und Milz: Eine Leberbeteiligung kann klinisch inapparent verlaufen.1 Bei bis zu 60% der Patienten kommt es jedoch zu Fieber, Nachtschweiß, Anorexie und Gewichtsverlust.1 Eine Hepatomegalie ist selten.2
- Neurologische Manifestation: Ca. 10% aller Patienten präsentieren sich mit neurologischen Auffälligkeiten.1 Klinische Symptome umfassen Lähmungen der Hirnnerven, Kopfschmerzen, Ataxie, kognitive Dysfunktion, Schwäche und Krampfanfälle.1 Eine aseptische Beteiligung der Meningen kann Symptome wie Fieber und Kopfschmerzen hervorrufen aber auch asymptomatisch verlaufen.2 Das Heerfordt Syndrom, das mit Uveitis, Parotisvergrößerung, Fieber und kranialer Neuropathie (Vorzugsweise Lähmung des VII Hirnnerven) einhergeht, ist sehr suggestiv für Sarkoidose.2
- Manifestation an den Augen: Eine Beteiligung des Auges liegt je nach anatomischen Befall in 25-80% der Fälle vor.1 Dabei kann jeder Teil des Auges betroffen sein.2 Eine Uveitis ist die häufigste Manifestation und tritt bei 65% der Patienten auf.1 Eine chronische anteriore Uveitis mit die mit Glaukomentwickung und Visusverlusts einhergehen kann, ist dabei häufiger als eine akute anteriore Uveitis.1 Bei ca. 10-15% der Patienten sind anteriores und posteriores Segment beteiligt.1 Die Konjunktiva ist in 6-40% der Fälle betroffen.2 Eine Beteiligung der Tränendrüsen kann zur Siccakeratokonjunktivitis führen.2
- Herzbeteiligung: Kardiale Granulome finden sich in bis zu 25% bei autopsierten Patienten.1 Klinisch apparent wird eine kardiale Beteiligung jedoch nur in ca. 5% der Fälle.1 Bevorzugte Lokalisationen der Granulome beinhalten den linken Ventrikel und das intraventrikluläre Septum.1 Klinische Manifestationen bei einer kardialen Beteiligung umfassen Blockbilder wie ein AV-Block2 und ein kompletter Rechhtschenkelblock2 aber auch Rhythmusstörungen wie eine Bradyarrhythmie oder paroxysmale Tachykardien,6 die mit Palpitationen, Synkopen und dem plötzlichen Herztod einhergehen können, seltener auch eine Kardiomyopathie.1
- Hyperkalzämie und Nierenbeteiligung: Zur Hyperkalzurie kommt es bei 40% der Patienten, zur Hypercalziämie bei 11% der Patienten.1 Eine intrarenale Kalziumablagerung kann zum Nierenversagen führen.1 Weiterhin kommt es im Rahmen der Sarkoidose zu einem gestörten Kalziumstoffwechsel.1 Ein Nierenversagen aufgrund einer granulomatösen Nephritis ist hingegen selten.1
- Knochen und Gelenkbeteiligung: In den meisten Fällen wird sie von den Patienten nicht wahrgenommen, kann jedoch auch zu Schmerzen führen. Chronische Arthralgien1 sowie eine Beteiligung der kleinen Gelenke sind häufig.2
- Weitere Organmanifestationen: Sie umfassen den Gastrointestinaltrakt, die Ohren und die Speicheldrüsen.2
Diagnostik
- Labor: Im akuten Stadium findet sich in aller Regel eine Erhöhung der BSG.9 Daneben kommt es in über 50% d. F. zu einer Erhöhung der Gammaglobuline.9 Bei Nierenbeteiligung kann eine Hyperkalzurie und Hyperkalzämie, bei Leberbeteiligung eine Erhöhung der Cholestaseparameter vorliegen.9 Angiotensin-converting enzyme (ACE) wird in den Epitheloidzellen der Granulome gebildet, und führt bei ca. 60% der Patienten zu erhöhten ACE-Serumkonzentrationen.1 Dennoch handelt es sich hierbei um einen unspezifischen und nicht besonders sensitiven Parameter, der aus diesen Gründen zum therapeutischen Monitoring primär nicht empfohlen wird.1
- Röntgen Thorax: Die Sarkoidose wird röntgenologisch in vier Schweregrade eingeteilt.1,2 Die radiologischen Stadien beziehen sich allein auf die Morphologie (Lymphadenopathie, und Lungenbeteiligung mit oder ohne Fibrose2 ) und korrelieren nicht mit der Lungenfunktion.1
- Grad 1: Bilaterale hiliäre symmetrische und nicht verdrängende Lymphadenopathie2 Dieses Stadium ist reversibel.9
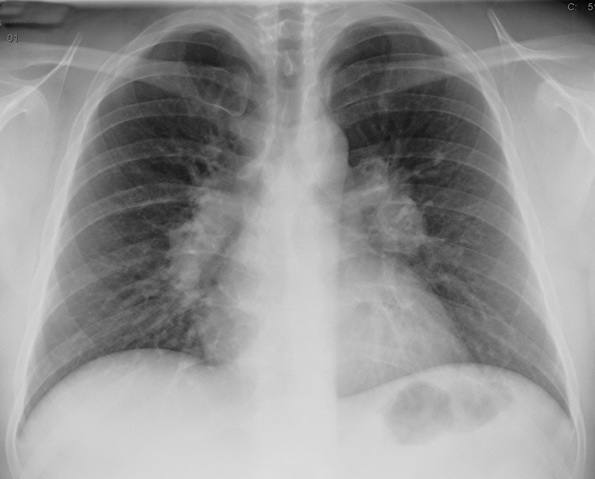
Abb. 481: Röntgen Thorax einer 50. jährigen Patientin. Erkennbar ist eine bilaterale hiläre nicht verdrängende Lymphadenopathie. Bei histologisch gesicherter Sarkoidose entspricht der radiologische Befund einer Sarkoidose Grad I. Quelle: www.rad.rwth-aachen.de/lernprogramm/rh_f.htm_
- Grad 2: Bilaterale hiliäre Lymphadenopathie mit Lungenbeteiligung.1Letztere tritt meist beidseitig, symmetrisch und diffus in Erscheinung mit Bevorzugung der zentralen Regionen und des Oberlappen.2 Erkennbar ist eine mikro-noduläre Infiltration, die mit diffusen punktförmigen Trübungen einhergeht.2
- Grad 3: Lungenbeteiligung ohne Lymphknotenvergrößerung.
- Grad 4: Lungenfibrose, Bullae und Bronchiektasien. Dieses Stadium ist Irreversibel.9
Am einfachsten erfolgt die radiologische Diagnose der Sarkoidose bei Grad I und II.
- Bronchioalveoläre Lavage: In 90% der Fälle findet sich eine Lymphozytose mit einem erhöhten CD 4/CD 8 Quotienten2 von normal 1,8:1 auf über 3,5.7 Bei starker fibrotischer Aktivität kann zudem die Neutrophilen- oder Eosinophilenzahl vermehrt sein.6 Daneben dient die BAL auch zum Ausschluss einer infektiösen Ursache und kann zur Beurteilung der Aktivität der Erkrankung beitragen.6 Weiterhin sollte im Rahmen der Routinediagnostik immer eine Kultur auf Mycobacterium tuberculosis und Pilze angelegt werden.6 Eine alleinige BAL ist zur Diagnose der Sarkoidose nicht ausreichend.3
- Bronchoskopie mit Lungenbiopsie: Sie ist zu Zeit noch Goldstandard und indiziert bei allen Patienten mit Ausnahme bei Löfgren-Syndrom1,3 Die Treffsicherheit bei mukosaler und/oder transbronchialer Biopsie liegt bei ca. 57-88%.2 Liegt kein eindeutiges Ergebnis vor (bei ca. 1/3 der Fälle3 ), so folgen weitere Untersuchungen.1 Die Biopsie dient neben der Festigung der Diagnose weiterhin dem Ausschluss einer malignen Erkrankung, einer Tuberkulose und einer Infektion mit Histoplasmen.3
- Mediastinoskopie: Bei unzureichenden Ergebnissen der oben genannter Biopsie sollte eine Mediastinoskopie mit Probenentnahme der mediastinalen Lymphknoten erfolgen.1 Komplikationen können in 2-3% der Fälle auftreten.
- EUS-FNA und EBUS-TBNA: Endoscopic ultrasound-guided transbronchial fine-needle aspiration und endobronchial ultrasound guided transbronchial fine-needle aspiration sind neue Verfahren in der Diagnostik der Sarkoidose und ermöglichen eine endoskopische Echtzeitbetrachtung und –biopsie verdächtiger Lymphknoten.3 Mit der EUS-FNA lassen sich besonders gut die linksseitigen unteren mediastinalen und paraösophagealen Lymphknotenstationen, mit der EBUS-TBNA die paratrachealen, subcarinalen und hilären Lymphknotenstationen darstellen.3
- 67 Ga-Citrat Szintigrafie: 67 Ga ist in der Diagnostik der Sarkoidose weit verbreitet.4 Es akkumuliert vor allem in Bereichen mit erhöhter Durchblutung, also vorzugsweise in entzündetem Gewebe.4 In der 67 Gallium-Szintigrafie gelten das sogenannte Panda-Zeichen (Isoptopenspeicherung in Parotis und Tränendrüse; ergibt das Bild ähnlich dem Gesicht eines Pandabären) und das Lambda Zeichen (Darstellung der mediastinalen Lymphknoten in Form eines Lamda) als charakteristisch für das Vorliegen einer Sarkoidose.4 Das gleichzeitige Auftreten beider Zeichen ist sehr spezifisch für das Vorliegen einer Sarkoidose.4
- FDG-PET: FDG-uptake im PET bei Patienten mit Sarkoidose ist unspezifisch und kann auch in malignen Erkrankungen wie Lymphomen oder Metastasen in Erscheinung treten.4 Aus diesem Grund ist die FDG-PET zur alleinigen Diagnosestellung ungeeignet.4 Dennoch kann das Verfahren zum therapeutischen Monitoring gut verwendet werden.4 Auch wenn die FDG-PET alleine als unspezifisch gilt, so eignet sie sich vor allem in Kombination mit der CT gut für den Krankheitsverlauf.4
- CT: Ein CT dient der weiterführenden Diagnostik und kann indiziert sein, wenn das Röntgenbild untypisch für eine Sarkoidose ist, oder Hämoptysen vorliegen.1 Typische Veränderungen umfassen perlschnurartige Reihungen von Noduli entlang der bronchovaskulären Bündel, der interlobulären Septen, am Lappenspalt und subpleural, die einer lymphatischen Ausbreitungsform entsprechen.6 Die Noduli können zu homogenen Verdichtungen mit flauen Rändern konfluieren, und so Einschmelzungen, Zysten oder ein Bronchopneumogramm imitieren.6 Weiterhin kann mit Hilfe eines CT in 5-15% eine Leber-, Milzbeteiligung und eine abdominelle Lymphadenopathie ausfindig gemacht werden.1
- Knochenszintigrafie: Sie dient dem Nachweis einer Knochenbeteiligung.4 Die Läsionen können entweder osteolytisch oder osteonekrotisch zur Darstellung kommen, und finden sich meist in den kleinen Gelenken.4 Zum Teil sind sie nur schwer von metastatischen Läsionen abzugrenzen.1
- MRT: Mit Hilfe des Schädel-MRT lässt sich eine Beteiligung des ZNS am besten erfassen.2 Ein besonderes Charakteristikum stellt die Granulombildung im Hypophysenstiel dar, die häufig zu Diabetes insipidus führt.6 Daneben wurden auch basale Meningitiden und insultartige Krankheitsbilder beschrieben.6 Sie dient neben der Diagnostik auch dem Therapiemonitoring.2
- Lungenfunktion: Ca. 65% der Patienten zeigen Einschränkungen in der Lungenfunktion.1 Für gewöhnlich liegt eine restriktive Ventilationsstörung vor, mit reduzierter CO Diffusionskapazität,2 VC und reduzierter FEV1.1 Bei 80% der Patienten kommt es im Rahmen der Ausheilung innerhalb von 2 Jahren wieder zu normalen Werten.1
Diagnose
Die Diagnose wird mit Hilfe der Klinik und der radiologischen Ergebnisse gestellt. Dies gilt vor allem für das Löfgren-Syndrom. Bei nicht ausreichenden Befunden kann die Histologie zur Rate gezogen werden.1
Differentialdiagnose
- Morbus Hodgkin
- Tuberkulose ,2 Hiliuslymphknoten Tbc ,9 atypische Mykobakteriose und Barthonelleninfekt6
- Berylliose2,5
- Sarcoid like lesions: Hierbei handelt es sich um lymphozytäre Entzündungen mit epitheloidzellhaltigen Granulomen, die sich histologisch nicht von der Sarkoidose abgegrenzt lassen. Sie können im Rahmen der „common variable immunodeficiency“ (CVID), einer HIV-Erkrankung und bei Tumorerkrankungen auftreten.6
- M. Wegener2
- M. Crohn bei gastrointestinalem Befall2
- Pilzinfektionen in der Lunge2
Therapie
Ein Großteil der Patienten, (ca. 30-70%2 die an Sarkoidose erkrankt sind, zeigen keine Beeinträchtigung durch die Krankheit und benötigen keine Therapie.1
Die Basis für die Therapie der Sarkoidose bilden systemisch verabreichte Glucocorticoide.2 Sie hemmen die Bildung von Granulomen und sind vor allem bei Herz-, ZNS-, Nieren-, und Augenbeteiligung sowie bei Hyperkalziämie indiziert.2 Weiterhin sollten symptomatische Patienten mit pulmonaler Beteiligung und Grad II und III Manifestationen im Röntgen-Thorax einer Kortisontherapie zugeführt werden.2 Ein internationales Expertengremium schlägt zur initialen Therapie 20-40mg Prednison/d vor.1,5 Nach einem und drei Monaten sollte eine Therapiekontrolle erfolgen.1,5 Sprechen die Patienten auf die Therapie an, kann die Prednisondosis auf 5-15mg pro Tag reduziert werden.1 Mit dieser Dosis wird die Therapie dann für weitere 9-12 Monate fortgeführt.1 Die Dosis darf jedoch nicht zu schnell reduziert werden.1 Gründe für ein mögliches Therapieversagen nach drei Monaten beinhalten unter anderem eine bereits eingetretene Lungenfibrose (Grad IV im Röntgen Thorax), eine mangelnde Compliance oder eine zu niedrige Dosis an Prednison.1
Als weiteres Medikament kann neben dem Prednison noch Methotrexat (10mg/Woche5 ) zum Einsatz kommen.1 Es findet vor allem dann Verwendung, wenn Steroide alleine keine ausreichende Wirkung zeigen2 und kann helfen diese einzusparen, vor allem dann, wenn absehbar ist, dass eine Therapie über einen längeren Zeitraum erfolgen muss.1,5 Azathioprin kann wie MTX zum Einsparen von Steroiden verwendet werden.2
Bei einem Erythema nodosum kann eine nichtsteroidale antientzündliche Therapie oder Colchizin über mehrere Wochen ausreichend sein.2 Hydroxychloroquin kommt vor allem bei Hypercalciämie, Hautbeteiligung und ZNS-Befall zum Einsatz.1 Cyclophosphamid kommt gerne bei kardialer und neurologischer Beteiligung zum Einsatz, vor allem dann, wenn Cortison nicht ausreicht und Thalidomid wird erfolgreich bei Lupus pernio eingesetzt.2
TNF-α Inhibitoren wie Infliximab sind neue Medikamente und haben in kleinen Fallstudien bei therapierefraktärem Lupus pernio, Uveitis und ZNS Befall eine gute Wirksamkeit gezeigt.5 Die Gabe von Infliximab kann mit Cortison kombiniert werden.6 Allerdings ist dieses Präparat trotz erwiesener Wirksamkeit im Rahmen einer internationale Multicenterstudie bisher nicht für die Sarkoidose zugelassen.6Die Studie hatte zwar gezeigt, dass die zusätzliche Gabe von Infliximab zu Prednisolon neben einer lungenfunktionellen Verbesserung auch zu einer Verbesserung der kutanen Sarkoidose führt, dennoch wurde der primäre Endpunkt nicht erreicht, sodass von Seiten der FDA weitere Studien eingefordert wurden, welche die herstellende Firma jedoch abgelehnt hat.6 Dies hat zur Folge, dass die Kostenübernahme für jeden Patienten im Einzelfall mit der zuständigen Krankenkasse verhandelt werden muss.6 Etanercept ein weiterer TNF-α Inhibitor konnte hingegen bei progredienten Lungenbefall mit dem Endpunkt Dyspnoe, Lungenfunktion und Röntgen Thorax keinen Therapieeffekt zeigen.5
Im Endstadium der Erkrankung kann eine Lungen-, Leber- und Herztransplantationen in Erwägung gezogen werden.2
Empfehlungen und symptomatische Behandlung bei Patienten mit Sarkoidose umfassen eine kalziumarme Diät, meiden von Sonnenlicht bei Hautbeteiligung, Sauerstofftherapie bei Lungenfibrose im Endstadium, Schrittmacher und ICD Implantation bei Rhythmusstörungen, und Antiepileptika bei Krampfanfällen.2
Komplikationen
- Pulmonale Hypertension: Sie ist eine mögliche Komplikation der Sarkoidose.1 Dem zu Grunde liegt in der Regel die Lungenfibrose.1 In seltenen Fällen kann auch die Infiltration der kleinen pulmonalen Arterien mit Granulomen zur Pulmonalen Hypertonie führen.1
- Respiratorisches Versagen: Auch hier ist die Ursache eine Lungenfibrose.2
- Portale Hypertension und Hepatopulmonales Syndrom: Bei ca. 1% aller Patienten kann es im Verlauf der Erkrankung über eine portale Hypertension und einem hepatopulmonalem Syndrom mit refraktärer Hypoxämie und Zirrhose zu einem Leberversagen kommen.1,2 Weiterhin kann sich eine chronische intrahepatische Cholestase entwickeln.2
- Plötzlicher Herztod: Er kann im Rahmen einer VT auftreten.2
Prognose
Zur spontanen Ausheilung kommt es bei der akuten Sarkoidose in mehr als 95% der Fälle innerhalb der ersten zwei Jahre.9 Die Spontanheilungsrate bei der chronische Sarkoidose beträgt ca. 70% innerhalb der ersten 1-3 Jahre bei Grad I, bei Grad II beträgt sie nur noch 50% und bei Grad III nur noch ca. 20%.9 Bei einem Viertel der Patienten kann die Sarkoidose chronifizieren.9 Folgende Prognosefaktoren kennzeichnen einen ungünstigen Verlauf: farbige Patienten, Alter >40 Jahre, Lupus pernio, chronische Uveitis, sinunasale und/oder ossäre Beteiligung, Beteiligung des ZNS und/oder des Herzens, Grad III und IV im Röntgen Thorax, chronische Hyperkalziämie und eine Nephrokalzinose.2 Die Lungenfibrose stellt das Endstadium der Sarkoidose dar, und ist die häufigste Ursache für Morbidität und Mortalität im Westen Europas.2 Ca. 20-25% der Patienten entwickeln im Verlauf eine Lungenfibrose.1 Die Mortalität beträgt in etwa 0,5-5% und ist meist Folge einer kardialen Beteiligung, eines ZNS-Befalls oder eines respiratorischen Versagens.1,2 Ein Rückfall ein Jahr nach eingetretener Remission ist selten und betrifft in der Regel weniger als 5% der Patienten.1
Weiterführende Literatur
Medical Progress Sarcoidosis
Michael C. Iannuzzi, M.D., Benjamin A. Rybicki, Ph.D., and Alvin S. Teirstein, M.D.; Medical Progress Sarcoidosis; N Engl J Med 2007; 357:2153-65.Sarcoidosis (Orphanet Journal of Rare Diseases)
Hilario Nunes, Diane Bouvry, Paul Soler2 and Dominique Valeyre; Sarcoidosis; Orphanet Journal of Rare Diseases 2007, 2:46Sarkoidose (Internist)
Prasse A, Müller-Quernheim J: Sarkoidose. Internist 50: 581–590, 2009
Referenzen
1 Michael C. Iannuzzi, M.D., Benjamin A. Rybicki, Ph.D., and Alvin S. Teirstein, M.D.; Medical Progress Sarcoidosis; N Engl J Med 2007; 357:2153-65.
2 Hilario Nunes, Diane Bouvry, Paul Soler2 and Dominique Valeyre; Sarcoidosis; Orphanet Journal of Rare Diseases 2007, 2:46
3 Skaidrius Miliauskas, Marius Žemaitis, Raimundas Sakalauskas; Sarcoidosis – moving to the new standard of diagnosis? ; Medicina (Kaunas) 2010; 46(7):443-6
4 Hima B. Prabhakar, Chad B. Rabinowitz, Fiona K. Gibbons, Walter J. O’Donnell, Jo-Anne O. Shepard, and Suzanne L. Aquino; Imaging Features of Sarcoidosis on MDCT, FDG PET, and PET/CT; AJR 2008; 190:S1–S6
5 Vincent Cottin; Update on bioagent therapy in sarcoidosis; F1000 Medicine Reports 2010, 2:13
6 A. Prasse · J. Müller-Quernheim; Sarkoidose; Internist 2009; 50:581–590
7 C. Thomas, Spezielle Pathologie; Schattauer Stuttgart-New York, 1996
8 W. Remmele, Pathologie Bd. 3, Springer-Verlag Berlin Heidelberg, 2. Auflage 1996
9 Gerd Herold und Mitarbeiter, Innere Medizin 2010
Lehrtexte Spezielle Pathologie

<< Respirationstrakt - Non-Tumor >>
Respirationstrakt - Non-Tumor - weitere Lehrtexte
Respirationstrakt - Non-Tumor - Kasuistiken
Organpathologie-Atlas
Weiterführende Literatur