Alveolarproteinose (PAP)
Definition
Bei der pulmonalen Alveolarproteinose handelt es sich um eine seltene Lungenerkrankung, die durch eine Anhäufung großer Mengen phospholipid- und proteinhaltiger Materialien in den Alveolen der Lungen gekennzeichnet ist.1,4,6 Die PAP lässt sich in eine kongenitale, sekundäre und idiopathische Form unterteilen.1,2Epidemiologie
Die PAP tritt mit einer Inzidenz von ca. 0,36/1000000 und einer Prävalenz von 3,7/1000000 auf, und hat zwei Häufigkeitsgipfel, zum einen in der Neonatalperiode (kongenitale PAP), sowie zwischen dem 30 und 40 Lebensjahr (idopatische und sekundäre PAP).4 Die idiopathische Form der PAP betrifft 9 von 10 Patienten und stellt damit die häufigste der drei Formen dar.1,2 Die sekundäre PAP tritt mit 5-10% etwas seltener auf,2 während die kongenitale PAP mit 3% eher zu den Raritäten zählt.2 Das Durchschnittsalter bei Diagnosestellung beträgt ca. 39 Jahre.1 Zwischen Symptombeginn und Diagnose vergehen laut Studien im Mittel bis zur sieben Monate.1,2 Die Erkrankung ist 2-3 mal häufiger bei Männern als bei Frauen, wobei ca. 72% der Patienten Raucher sind.1
Pathogenese
In den vergangenen Jahren konnten die der PAP zugrunde liegenden Prozesse zu einem großen Teil aufgeklärt werden. Dabei rückte GM-CSF, ein potenter Stimulator der myeloischen Hämatopoese in den Vordergrund der Forschungen.1
Lungenepithelzellen dienen als Quelle von GM-CSF.1 GM-CSF bindet an einen Rezeptor an der Zelloberfläche von Typ II Pneumozyten und Alveolarmakrophagen bestehend aus einer α-und einer β-Kette.1 Letztere ist zudem ein Bestandteil der Rezeptoren für IL-3 und IL-5.1 Knockout Mäuse, denen entweder das Gen für GM-CSF fehlte, oder eine selektive Deletion der gemeinsamen β-Kette des GM-CSF/ IL-3/IL-5-Rezeptors, die für eine Bindung von GM-CSF an Makrophagen erforderlich ist, aufzeigten, entwickelten eine Lungenerkrankung, die histologisch der humanen PAP glich,5 ohne dabei eine überschießende Hämatopoese zu entwickeln.1 Darüber hinaus zeigten die Mäuse auch einen Defekt in der Surfactant-Clearance, Anfälligkeit für Infektionen und eine beeinträchtigte Funktion der Alveolarmakrophagen.1
Es konnte in Rahmen von Studien gezeigt werden, dass eine intrapulmonale exogene GM-CSF Gabe sowie eine Überexpression von GM-CSF zur Besserung der Symptomatik führte.1
Bei der Mehrzahl der Patienten mit PAP ist der GM-CSF/ IL-3/IL-5-Rezeptor jedoch intakt, und die Expression des GM-CSF Gens sowie die GM-CSF-Produktion sind normal.5 Trotzdem ist die GM-CSF-Wirkung auf die Alveolarmakrophagen vermindert und ihre Funktion bei Patienten mit PAP gestört.5 Untersuchungen wiesen bei den betroffenen Patienen in der BAL-Flüssigkeit und im Serum einen Autoantikörper gegen GM-CSF nach.1,5 Dieser bindet an das biologisch aktive GM-CSF und neutralisiert dessen Wirkung.5 Das ausbleibende Signal des Zytokins beeinträchtigt einerseits die Surfactant-Clearance durch die Alveolarmakrophagen und andererseits deren Funktion bei der Abwehr von Infektionen.1,5 Folglich kann die idiopathische PAP als Autoimmunerkrankung eingestuft werden.1
Zusammenfassend konnte davon ausgegangen werden, dass dem Wachstumsfaktor GM-CSF eine wesentliche Rolle bei der Surfactant-Clearance in den Alveolen zukommt und eine reduzierte Bioverfügbarkeit des Zytokins an der Pathogenese der PAP beteiligt ist.5
Die kongenitale PAP ist hingegen auf eine hereditäre Defizienz von SP-B zurückzuführen,4 verursacht durch eine Frame-Shift-Mutation im Gen für SP-B 2.3
Bei der sekundären Form der PAP kommen neben externen Noxen auch eine Reihe von Erkrankungen als Auslöser in Frage.1 Ursachen für eine sekundäre PAP umfassen:
- Inhalative Noxen (Silikat, Asbestfasern, Molybdän)5
- Exposition gegenüber Insektiziden, Aluminiumstaub, Titanium-Dioxyd und anderen anorganischen Stäuben.4
- Hämatologische Erkrankungen, (vor allem myeloische Leukämie und MDS)
- HIV-Infektion (AIDS),1 Hypergammaglobulinämie und IgA-Mangel.4
- “Lysinuric protein intolerance“ (LPI; selten)1
- Infektionserkrankungen (Pneumocystis carinii, Mykobakterien, Nocardia, atypische Mykobakternien, Zytomegalievirus)5
- Amyloidose5
Makroskopie
Ein ungewöhnlich hohes Eigengewicht der Lunge, welches das Doppelte bis 5fache der Norm betragen kann, ist neben einer gelblichen, von milch- und eiterähnlicher Flüssigkeit bedeckten Schnittfläche die auffälligste makroskopische Veränderung. [6] Nach dem Abstreifen der Flüssigkeit kommen multiple gelblich-weiße Noduli, die meist nur wenige Millimeter messen, zur Darstellung.6
Mikroskopie
Histologisch findet sich intraalveolär, ein amorphes eosinophiles feingranuläres Material das reichlich Lipide enthält.2
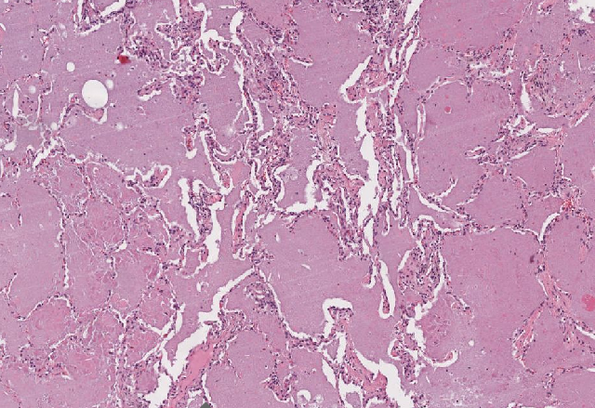
Abb. 484: Erkennbar ist ein amorphes eosinophiles feingranuläres Material in den Alveolen
Es kann auch in terminalen und respiratorischen Bronchiolen lokalisiert sein; im Interstitium hingegen lässt es sich nicht nachweisen.6 Das feingranuläre Material stellt sich in der PAS-Färbung rot und in der May-Grünwald-Giemsa Färbung basophil dar, bei Negativität mit Alcanblau.2 Mit Ausnahme der kongenitalen PAP lässt sich das feingranuläre intraalveoläre Material zusätzlich mit SP-A anfärben.
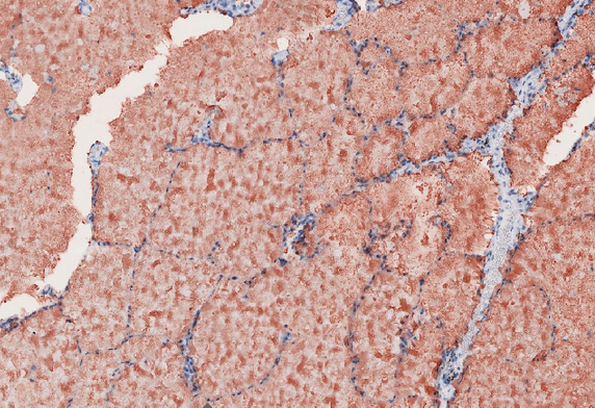
Abb. 485: Anfärbung des feingranulären intraalveolären Materials mit SP-A.
Weiterhin sind vor allem in der Lungenperipherie aufgeschwemmte und degenerativ veränderte Makrophagen mit schaumigen Zytoplasmaveränderungen nachweisbar.6 Die alveoläre Architektur ist in der Regel normal, manchmal finden sich Verdickungen des Interstitiums bedingt durch Ödembildung, eine lymphozytäre Infiltration oder Hyperplasie der Alveolarmakrophagen.1,2,4 Nur bei sehr langjährigen progredienten Verläufen lassen sich fibrotische Veränderungen nachweisen.1
Differentialdiagnose
Differentialdiagnostisch sollten folgende Erkrankungen in Erwägung gezogen und ausgeschlossen werden:
- Chronische Bronchitis1
- Lungenödem2
- Hypersensitivitätspneumonie3
- organisierte Pneumonie3
- Pneumozystis carinii Pneumonie (PCP)2,3
- bakterielle Pneumonie3
- interstitielle Pneumonitis3
- Bronchiolitis obliterans3
- ARDS3
Klinik
Die Symptome können schleichend über Jahre verlaufen und beinhalten neben dem Leitsymptom Dyspnoe, die meist erst unter Anstrengung auftritt, noch unproduktiven trockenen Husten, Unwohlsein,1 und leichtes Fieber.4 Gewichtsverlust, Fatique,2 Thoraxschmerzen2 und Hämoptysen können auftreten, sind aber eher selten.1
Der klinische Untersuchungsbefund ist meist unauffällig, manchmal finden sich inspiratorische Rasselgeräusche und eine Zyanose.2 Ein Drittel der Patienten weisen bei Diagnosestellung bereits Trommelschlegelfinger auf.4
Bei der kongenitalen PAP präsentiern sich die Kinder mit Atemnot, die sich bei Gabe von Surfactant und Kortikosteroiden nicht bessert.1
Diagnostik
- Labor. Bei der idiopatischen Form spielt der Nachweis von Autoantikörpern gegen GM-CSF eine wichtige Rolle.1,2,4 Bei Patienten mit sekundärer oder kongenitaler PAP sind hingegen keine Auto-AK nachweisbar.4 Die Titer korrelieren allerdings nicht mit dem Krankheitsgeschehen.2 Da auch 0,3-2% der gesunden Erwachsen niedrigtitrige Auto-AK aufweisen und mit GM-CSF therapierte Patienten ebenfalls Auto-AK bilden können,4 eignet sich der Auto-AK Nachweis gegen GM-CSF nur bedingt für die alleinige Diagnosestellung. Weitere nicht spezifische serologische Veränderungen umfassen eine Erhöhung der LDH, deren Konzentration im Gegensatz zu GM-CSF mit der Krankheitsaktivität korreliert5, der Tumormarker CEA und dem muzinähnlichen Glykoprotein (KL-6), sowie der Surfactantproteine SP-A und SP-D.4 Nach therapeutischen Lavagen kann es zur Normalisierung der genannten Parameter kommen.4
- BGA: Leitsypmtom ist eine Hypoxämie, die insbesondere unter Anstrengung auftritt.4 Die O2 Sättigung ist erniedrigt.4 Der pCO2 liegt meist im Normbereich.4
- Lungenfunktion: Primär liegt eine restriktive Ventilationssstörung vor, mit Reduktion der Vital- und Totalkapazität sowie der CO-Diffusionskapazität.1,2 Daneben findet sich ein erhöhter alveolar-arterieller Sauerstoffgradient (AaDPO2) und eine gesteigerte pulmonale Shuntfraktion.5
- Röntgen-Thorax: Im konventionellen Röntgen finden sich in mehr als 50% der Fälle bilaterale symmetrische perihiläre alveoläre Infiltrate mit einer flügelartigen Verteilung, auch “bat´s wing sign“ genannt.4 Apikale Lungenabschnitte und die costodiaphramalen Winkel bleiben meist ausgespart.2 Die Infitrate können zur Milchglastrübung (Ground-glass-pattern), lineraren, retikulären oder nodulären Verdichtungen und Verschattungen führen.4 Es können aber auch multifokale asymmetrische Infiltrationen auftreten.2 Lymphknotenvergrößerungen,4 Pleuraergüsse, Lungenödeme oder eine Kardiomegalie finden sich in der Regel nicht.2,5
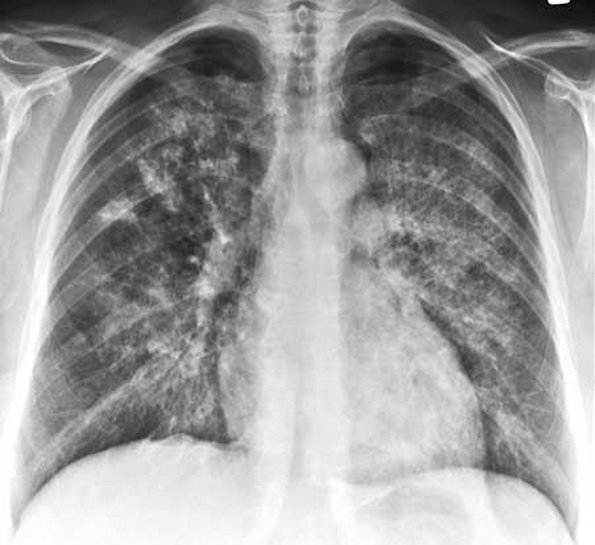
Abb. 483: Röntgen Thorax, mit bilateralen symmetrischen retikulonodulären Verschattungen beider Lungen mit Aussparung der Hili. Quelle: www.mevis-research.de.
- HR-CT: Charakteristisch für die adulte PAP sind “crazy paving pattern“.2,4 Hierbei handelt es sich um milchglasartige Trübungen mit darüber gelegenen interlobären septalen Verdickungen und intralobulären interstitiellen Verdichtungen.1,2,4 Diese Veränderungen sind hoch charakteristisch für eine PAP, treten aber auch bei Infektionen und Blutungen u.a. auf.2
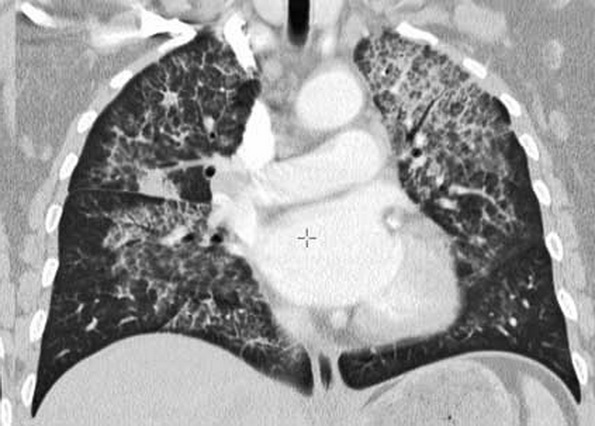
Abb. 482: CT-Thorax, Lungenfenster, koronare Aufnahme. Erkennbar sind Areale mit “crazy paving pattern" und mit Aussparung der costodiaphramalen Winkel. Quelle: www.mevis-research.de._
- BAL: Makroskopisch fällt eine milchig trübe Flüssigkeit auf.1,4 Lässt man die Flüssigkeit eine Weile stehen, kommt es am Boden zur Bildung eines trüben eingedickten Sediments, mit einem mit einem hellgelben fast durchscheinenden Überstand.1 Die biochemische Untersuchung der Lungenspülflüssigkeit kann entscheidende Hinweise auf das Vorliegen einer PAP geben.4 Bei der konnatalen Form ist das Fehlen von SP-B und die Anwesenheit von pro SP-C charakteristisch für den angeborenen SP-B-Mangel.4
- Lungenbiopsie: Reichen die genannten Untersuchungen zur definitiven Diagnose nicht aus, sollte eine Lungenbiopsie zur histologischen und immunhistologischen Diagnostik durchgeführt werden.4 Anhäufungen von intraalveolärem Periodic-Acid-Schiff-(PAS-) positivem Material sind typisch, entscheidend ist jedoch der fehlende Nachweis von SP-B und das Vorhandensein von pro-SP-C im Präparat.4
Diagnose
Die Kombination aus Klinik, Bildgebung und BAL ist in der Regel ausreichend, um die Diagnose PAP zu stellen.1
Therapie
Therapie der Wahl ist nach wie vor noch die mechanische Entfernung der phospholipid- und proteinhaltigen Materialien mit Hilfe der kompletten Lungenlavage (whole lung lavage, WLL).1 Bei der WLL werden die Lungen solange mit 0,9% NaCl Lösung gespült, bis das Bronchialsekret nicht mehr trüb ist.1 Der Vorgang kann zwischen 2-4h dauern und erfolgt in Vollnarkose.1 Patienten mit idiopatischer PAP profitieren am meisten von dieser Behandlung.1 Nach durchgeführter WLL tritt eine sofortige Besserung der klinischen Symptomatik, der radiologischen Befunde, sowie der Lungenfunktion mit einem Anstieg des PO2, der VC, FEV1 und CO-Diffusionskapazität ein.1 Das mittlere symptomfreie Intervall nach WLL beträgt ca. 15 Monate, kann aber von Patient zu Patient variieren.1,2 Manche Patienten benötigen im Leben nur eine WLL um beschwerdefrei zu bleiben.1 Nach zwei durchgeführten WLL erreichen ca. 60% der Patienten eine verbesserte körperliche Belastung.2 Eine kleine Zahl der Patienten profitiert jedoch nicht von der Therapie.1 Dabei gilt, je jünger die Patienten, desto wahrscheinlicher ein schlechteres Therapieansprechen.1
Bei der kongenitalen Form der PAP ist eine WLL , die Gabe von Surfactant3 und Cortison sowie die Gabe von GM-CSF wirkungslos.1 Hier kann nur eine frühe Lungentransplantation das Leben des Kindes retten.1,3 Die Kinder müssen fast immer invasiv beatmet werden, eine Alternative bietet die ECMO (extracorporal membrane oxygenation).3
In Ausnahmefällen kann eine GM-CSF-Applikation bei ausgewählten Patienten Linderung verschaffen.1 Obwohl die Gabe von GM-CSF im Allgemeinen gut vertragen wird, ist die Ansprechrate in Rahmen von zwei kleineren Studien mit weniger als 50% noch sehr enttäuschend, und damit nicht so effektiv wie die WLL.1,2
Tritt die PAP als Sekundärerkrankung in Erscheinung, sollte die zu Grunde liegende Erkrankung therapiert werden.1 Besteht eine Exposition gegenüber inhalativen Noxen, so sollten diese gemieden werden.1 Neben der Behandlung der Ursache, kann bei Ausbleiben einer Besserung auch noch eine WLL zum Einsatz kommen.1 Eine GM-CSF- Behandlung ist bei der sekundären PAP nicht gerechtfertigt, da bei den Patienten keine Antikörper nachweisbar sind.1
Prognose
Die Prognose der kongenitalen PAP ist deutlich schlechter als die der adulten Formen.1 Bei der idiopathischen PAP werden 5 und 10-Jahres Überlebensraten von 75% und 68% angegeben.1 Der einzig negative prognostische Faktor bei der PAP ist ein Alter unter 5 Jahren. Die 5-Jahres Überlebensrate nach Diagnosestellung beträgt hier nur 14%.1 Die Mortalitätsrate bei konventioneller Therapie sogar 100% 3 80% der Todesfälle treten in den ersten 12 Monaten ein.3 Die Lungentransplantation ist die einzige Aussicht auf Überleben.3 Zu einer erhöhten Mortaltität kommt es aber auch in einem Alter > 39 Jahre, diese ist aber nicht kankheitsspezifisch.1 Es ist anzunehmen, dass hierfür u.a. komplizierende Infektionen verantwortlich sind. Obwohl Raucher ein höheres Risiko haben, an der PAP zu erkranken, hat diese inhalative Noxe keinen Einfluss auf das Überleben.1
Bei einem langjährigen progredienten Verlauf der Erkrankung kann sich im weiteren Verlauf eine Lungenfibrose entwickeln.1 Versterben Patienten direkt an den Folgen der PAP, so umfassen die beiden häufigsten Todesursachen respiratorisches Versagen (80%) und Pneumonie (20%).2 Das Risiko im Rahmen einer PAP am respiratorischen Versagen zu sterben beträgt in etwa 10-15%1
Nur in wenigen Fällen kann es zu einer spontanen Remission der Erkrankung kommen.1
Weiterführende Literatur
Rare lung diseases II: Pulmonary alveolar proteinosis
S.C. Juvet MD, D.Hwang MD PhD, T.K. Waddell MD PhD, G.P. Downey MD, Rare lung diseases II: Pulmonary alveolar proteinosis, Can Respir 2008;15(4):203-210From the Archives of the AFIP Pulmonary Alveolar Proteinosis
Aletta Ann Frazier, MD et al.,From the Archives of the AFIP Pulmonary Alveolar Proteinosis, RadioGraphics 2008; 28:883–899Aktuelle Entwicklungen in der Pneumologie
Kroegel C, Reissig A, Bonnet R, Albes JM, Thole H, Ollenschläger G, Wahlers T, Schneider CP, Gillissen A, Costabel U: Aktuelle Entwicklungen in der Pneumologie 2002 – Teil 1. Med Klin 98: 30-56, 2003
Referenzen
1 S.C. Juvet MD, D.Hwang MD PhD, T.K. Waddell MD PhD, G.P. Downey MD, Rare lung diseases II: Pulmonary alveolar proteinosis, Can Respir 2008;15(4):203-210
2 Aletta Ann Frazier, MD et al.,From the Archives of the AFIP Pulmonary Alveolar Proteinosis, RadioGraphics 2008; 28:883–899
3 G. Garg, A. Sachdev, D. Gupta, Pulmonary Alveolar Proteinosis, Case Report, Indian Pediatrics, Volume 46 June 17, 2009
4 M. Griese, M. Tredano, M. Bahuau, Pulmonale Alveolarproteinose Molekulare Grundlagen und Konsequenzen für Diagnostik und Therapie, Monatsschr Kinderheilkd 2001, 149:1245-1261 Springer Verlag
5 C. Kroegel et al., Aktuelle Entwicklungen in der Pneumologie 2002-Teil 1, Med Klin 2003; 98:30–56.
6 J. Freyschmidt, M. Galanski, Handbuch diagnostische Radiologie, Bd. 4 Thorax, 2003 Springer Verlag Berlin Heidelberg New York
Lehrtexte Spezielle Pathologie

<< Respirationstrakt - Non-Tumor >>
Bilder
Abb. 484: Erkennbar ist ein amorphes eosinophiles feingranuläres Material in den Alveolen
Abb. 485: Anfärbung des feingranulären intraalveolären Materials mit SP-A.
Respirationstrakt - Non-Tumor - weitere Lehrtexte
Respirationstrakt - Non-Tumor - Kasuistiken
Organpathologie-Atlas
Weiterführende Literatur