M. Wegener
Definition
Beim Morbus Wegener handelt es sich nach der Chapel-Hill Konsensus-Konferenz von 1992 um eine granulomatöse Entzündung der Atemwege mit nekrotisierender Vaskulitis, die vorwiegend die kleinen bis mittelgroßen Gefäße (Kapillaren, Venolen, Arteriolen und Arterien) betrifft, und mit ulzerierenden, nicht verkäsenden Granulomen im Bereich des oberen und unteren Respirationstraktes (Nase, Nasennebenhöhlen, Mittelohr, Oropharynx und Lunge) sowie mit einer Beteiligung der Nieren (Glomerulonephritis (RPGN3 ), Mikroaneurysmen) einhergeht.2,6 Die Erkrankung kann sich auf alle Organsysteme des Körpers ausweiten.2 Ca. 2/3 der Patienten zeigen bei der Erstmanifestation eine pulmonale Beteiligung, etwas mehr als ein Drittel präsentieren sich mit einer RPGN.3,5 Davon entwickeln aber zwischen 80-90% der Patienten im weiteren Verlauf eine Nierenbeteiligung5.Die Wegener Granulomatose ist von den ANCA-assoziierten Vaskulitiden am häufigsten5.Epidemiologie
M. Wegener kann Patienten unterschiedlichen Alters betreffen, findet sich jedoch gehäuft im 5. Lebensjahrzehnt4. Männer sind etwas häufiger betroffen als Frauen4. Die Prävalenz beträgt in etwa 5/100000, die Inzidenz 0,9/100000/Jahr10Pathophysiologie
Die granulomatöse Histopathologie und der Nachweis eines dominierenden TH-1-Zytokinmusters der pulmonalen Lymphozyten sprechen für eine zelluläre Immunreaktion (Typ IV-Reaktion nach Gell und Coombs)9. Die Entstehung der Granulome beruht auf der Aktivität von T-Zellen und T-Zellzytokinen1. Der Erkrankungsbeginn im Respirationstrakt legt die Beteiligung eines exogenen Agens nahe. Es ist nachgewiesen, dass die Besiedlung der Schleimhäute durch Staph. aureus mit einer erhöhten lokalen Krankheitsaktivität und einer erhöhten Rezidivrate einhergeht, und eine Reduktion der Rezidivhäufigkeit durch eine antiinfektive Therapie erzielt werden kann9.
Im vaskulitischen Generalisationsstadium stellen die Neutrophilen die zentrale Effektorzellpopulation dar. Die Bindung von PR3-ANCA an die auf der Neutrophilenoberfläche exprimierte PR3 zur Zellaktivierung und Initiierung einer neutrophilenabhängigen Entzündungsreaktion ist führend. Folge sind Endothel- und Gefäßschäden1. Sowohl in der Lunge aus auch in der Niere wurden lokale Neutrophilendegranulationen bei aktiver Erkrankung und eine Korrelation zwischen Krankheitsaktivität und Neutrophilenaktivierung nachgewiesen. Der ANCA-Serumtiter korreliert in der Mehrzahl der Fälle mit der Krankheitsaktivität9.
Makroskopie
In den Lungen finden sich multiple, im Durchmesser bis 2cm große Knoten, mit entweder solidem Aufbau oder zentral kavernenartig zerfallen. Die große Variabilität pulmonaler Manifestationen bei der WG ergibt sich aus dem dualen Charakter dieser Erkrankung. Granulomatöse extravaskuläre Manifestationen im unteren Respirationstrakt präsentieren sich zum einen als Lungenrundherde aufgrund intraparenchymaler Läsionen oder als interstitielle Mikrogranulome, während sich der Befall der unteren Atemwege als ulzerierende Tracheobronchitiden und entzündliche, häufig stenosierende Pseudotumoren des Bronchialsystems manifestiert7.
Mikroskopie
Pathohistologisch ist der M. Wegener durch eine nekrotisierende granulomatöse Entzündung der kleinen Gefäßwände gekennzeichnet.
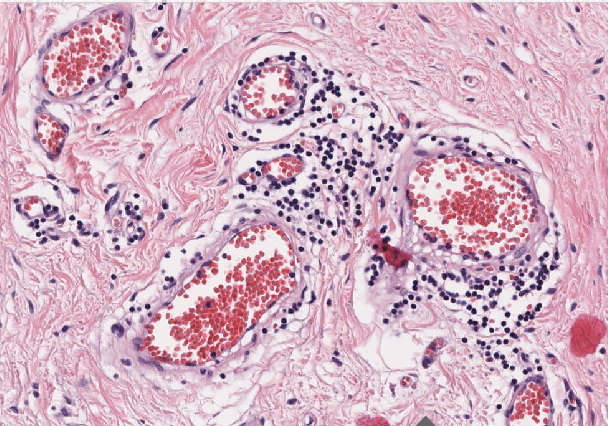
Abb. 523: Pulmonale Vaskulitis mit entzündlichen Infiltraten
Diese geht innerhalb der Lungen einher mit multiplen Nekrosearealen umgeben von Einblutungen, kleinen Mikroabszessen, bronchoalveoläre Entzündungen12, Fibrosen12 und Granulomen4. Die Parenchymnekrosen können als Mikroabszesse (65% der Fälle) oder als landkartenartige Nekrosen (85% der Fälle) vorkommen12. Fibrinoide Nekrosen werden von palisadenartig gestellten Histiozyten und von Riesenzellen begrenzt12. Morphologisch entwickeln sich die zur Nekrose neigenden Granulome aus Epitheloidzellen, Riesenzellen des Fremdkörper oder Langhans-Typs, Lymphozyten, Plasmazellen und Eosinophilen11.
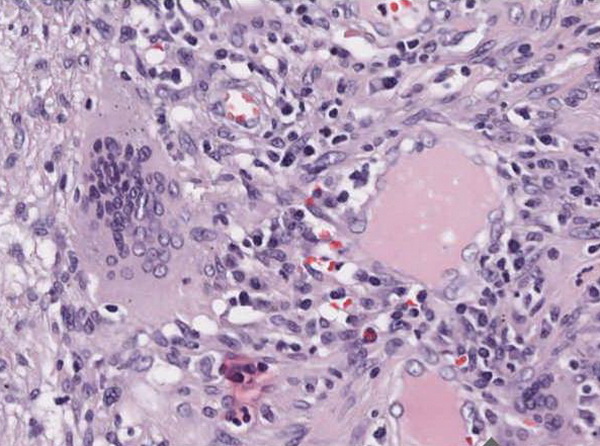
Abb. 443: Auf der linken Seite eine mehrkernige Riesenzelle, rechts daneben zwei kleine Nekrosen
An mittelern und keinen Arterien und kleinen Venen sind proliferative und nekrotisierende entzündliche Veränderungen nachweisbar. Auch die pulmonalen und bei Generalisation später auch in allen Organen auftretenden Herde sind ähnlich aufgebaut11. Entscheidend für die Differentialdiagnose gegenüber pulmonalen granulomatösen Infektionen ist der Nachweis der nekrotisierenden Vaskulitis, besonders der mittleren muskulären Arterienäste und Venen11.
Die WG stellt morphologisch insofern eine Besonderheit dar, als neben einer Vaskulitis – häufig dieser vorangehend – eine extravaskuläre entzündliche Komponente auftritt. Daher besteht die klassische histopathologische Trias der WG aus einer granulomatösen extravaskulären Entzündung mit landkartenartigen Nekrosen und einer (teils nekrotisierenden) Kleingefäßvaskulitis7.
Aufgrund der herdförmigen Verteilung der Gewebeläsionen müssen Biopsien gezielt aus läsionalem Gewebe entnommen werden. Da der obere Respirationstrakt bei der Mehrzahl der Patienten involviert ist, empfiehlt es sich, Biopsien aus der Nasenschleimhaut und den NNH zu entnehmen9. Es gilt bei Biopsieentnahme zu beachten, dass nicht alle Kriteren der WG mit nekrotisierenden Granulomen und Vaskulitis in einer Biopsie vollständig vorhanden sein müssen. Eine WG ist daher aufgrund einer negativen Biopsie nicht immer definitiv auszuschließen, vielfach gelingt die Diagnosestellung nur kumulativ aus mehreren Biopsien unter Einbeziehung der klinischen und serologischen Daten7
Bei der BAL zeigt sich eine lymphozytäre Alveolitis7.
Klinik
Klinisch unterscheiden sich zwei Stadien:2,3
1. Lokal begrenztes Initialstadium: Erkrankung des Respirationstraktes ohne Nierenbeteiligung und ohne systemische Vaskulitis.10 Hierbei handelt es sich um eine vorwiegend granulomatöse Form der Erkrankung, die gerne chronisch verläuft. [2] Sie betrifft ca. 1/3 der Patienten3. Das Stadium umfasst:
- Die oberen Atemwege sind bei 70-95% der Patienten betroffen. Sie umfassen eine chonische Rhinitis/Sinusitis einhergehend mit Nasenbluten und Krustenbildung, evtl. Sattelnase und Septumperforation2, evtl. auch Mastoidits10
- Ausbildung subglottischer- oder Glottispolypen, Larynx- oder Bronchialstenosen bedingt durch die granulomatöse Entzündung.2 Ulzerationen im Oropharynx10
- Pulmonale Beteiligung bei 70-95% der Patienten5. Lungenrundherde, evtl. mit zentralen Einschmelzungen, pulmonale Blutungen2 Ca. 90% der Patienten zeigen im Krankheitsverlauf eine pulmonale Beteiligung4. Diese zeigt sich klinisch mit Husten, Pleuritis, Dyspnoe, Hämoptysen4, sowie Thoraxschmerzen und tracheobronchiale und endobronchiale Infektionen in 10-50% der Fälle5.
- Epistaxis. chronische Otitis, und Hörverlust2.
2. Vaskulitisches Generalstadium mit möglichem pulmo-renalem Syndrom10 (rasch progredienter Verlauf der Erkrankung2 ): Sie betrifft ca. 2/3 der Patienten3
- Evtl. alveoläre Hämorrhagie mit Hämoptoe2
- fokale segmentale nekrotisierende Glomerulonepritris2. Klinisch manifestiert sich die Nierenbeteiligung als Hypertonus, Proteinurie, nephritisches und nephrotisches Syndrom bis hin zu den klinischen Zeichen des Nierenversagens. Die renale Beteiligung steigt mit zunehmenden Alter2, und tritt 50-90% der Patienten auf5.
- orbitale Beteiligung mit Uveitis und Ulcerationen in 25-50% der Fälle5, retinaler Vaskulitis mit Granulombildung2und Episkleritis10.
- Myalgien und Arthralgien und Arthritis treten bei ca. 50% der Patienten auf5
- ZNS-Symptome, periphere Neuropathien (Mononeuritis Multiplex)2
- Typisch ist eine Beteiligung der Haut mit klassischer tastbarer Purpura, Nodulae, Vesiculae5 und peripherer Gangrän mit Gewebedefekten2.
- Beteiligung von Herz (3-13%)5, Speicheldrüsen, Gonaden und Mamma2
- B-Symptomatik (Fieber, Gewichtsverlust, Nachtschweiß), allgemeines Unwohlsein, Wachstumsretardierung2
Beide Stadien können koexistieren2.
Diagnostik
- Labor: Oft BSG-Erhöhung4, Erythrozyturie, und Anstieg des Serumkreatinins (Glomerulonephritis) Leukozytose4, Thrombozytose, normozytäre Anämie4 und positive Rheumafaktoren4. Nachweis antineutrophiler cytoplasmatischer Antikörper, mit cytoplasmatischem Fluoreszenzmuster (cANCA) meist mit dem Zielantigen Proteinase 3 = Anti-Proteinase 3-Antikörper (PR3-ANCA) in der indirekten Immunfluoreszenz (IIF) und ELISA2,4.
c-ANCAs und Anti-PR3 ELISA haben eine Sensitivität zwischen 85-90% und eine Spezifität von 95% bei einer generalisierten aktiven Wegener Granulomatose mit niedriger Empfindlichkeit im lokal begrenzten Initialstadium (ca. 60%) und Erkrankung in Remission (ca.40%)5. pANCA mit Spezifität gegen MPO kann auch bei Patienten mit M. Wegener gefunden werden2. Falsch positive Befunde können bei anderen systemischen Autoimmunerkrankungen vorliegen wie z.B.: SLE und Rheumatoide Arthritis u.a.5. Alleinige Bestimmung der Titer im Rahmen der Rezidivdiagnostik sind nicht sensitiv und spezifisch genug, auch wenn ein Titeranstieg ein Hinweis auf ein Rezidiv gibt, sollte die Rezidivdiagnostik in erster Linie klinisch erfolgen5. Eine negativer ANCA-Status schließt eine WG nicht aus7. - Röntgen Thorax: In mehr als 80% der Fälle findet sich ein auffälliges Röntgen-Thorax5.Die häufigsten bildgebenden Befunde umfassen fokale alveoläre, interstitielle oder fokale Verschattungen5, die in Form und Größe variieren können mit max. Durchmesser von bis zu 10cm, i.d.R. aber 2-4cm4. Es bestehen keine besonderen Manifestationslokalisationen, die Verschattungen treten in der Regel multipel auf, haben eine rundliche bis ovale Form und führen, wenn größer als 2cm in 25% der Fälle zur Kavernenbildung4. Bei sekundärer Infektion findet sich Luft in den Höhlen4. Die Wände der Höhlen variieren in ihrer Dicke, werden aber bei fortbestehender Erkrankung mit der Zeit dünner4. Die Läsionen können mit oder ohne Narbenbildung abheilen4.
- HRCT: Noduläre Veränderungen und einschmelzende Infiltrate sind häufig bei Morbus Wegener. Noduli finden sich bei 55–70% und Kavernen bei 35– 50% aller Patienten mit WG8. Im HRCT können die Rundherde von einer Milchglasverschattung umgeben sein, welche Ursache einer Lungenblutung sein kann4. Subglottische Wandverdickung, Trachealwandverdickung, Bronchialwandverdickung und daraus resultierende Verengungen können zu einer sekundären Minderbelüftung der Lunge führen, und sind oftmals gegenüber systemischer Therapie resistent4. Von Bedeutung ist die differenzialdiagnostische Abgrenzung gegenüber Malignomen und infektiösen Prozessen (Tuberkulose)8.
Weiterhin können Pleuraverdickungen und keine Ergüsse mit der CT detektiert werden4. - Sonografie der Nieren: In einem frühen Stadium der Nierenbeteiligung finden sich meist große echogene Nieren im Ultraschall, die keine Anomalien im Doppler aufweisen4. Durch rasch fortschreitende Glomerulonephritis kann es innerhalb kurzer Zeit jedoch zu narbig veränderten Schrumpfnieren kommen4. Seltener kommt ein Pseudotumor zur Darstellung4.
- Bronchoskopie und Biopsie: Sie kommt bei Lungenblutung, endobronchialen Stenosen oder anderen Läsionen zum Einsatz und kann mit Hilfe der Biopsie aus suspekten Arealen zur Diagnosefindung beitragen5.
- Transbronchiale Biopsie: Sie sind im Großen und Ganzen unzureichend, um die Diagnose einer Vasculitis zu stellen5.
- VATS mit Lungenkeil: Besteht eine Beteiligung der Lunge, kann mit Hilfe der VATS und nachfolgender histologischer Aufarbeitung eines Lungenkeils die endgültige Diagnose eines M. Wegener sicher gestellt werden5.
- Nierenbiopsie: Die perkutane Nierenbiopsie wird häufig zur Diagnosestellung der akuten Glomerulonephritis durchgeführt5. Während histologische Charakteristika des M. Wegener wie granulomatöse Entzündungen oder Nekrosen in Gefäßen eher selten sind, kann die Diagnose der systemischen Vaskulitis durch Feststellung einer segmentalen nekrotisierenden Glomerulonephritis ohne Immunkomplexablagerungen (= pauci immuneGlomerulonephritis) in den meisten Fällen gestellt werden5. Es ist sehr wichtig, dass zusätzlich zur konventionellen Histopathologie, eine Immunfluoreszenz und eine elektronenmikroskopische Untersuchung der Proben durchgeführt werden5. Durch das Fehlen der charakteristischen Immunfluoreszenzmuster wie IgA Ablagerungen bei Patienten mit Henoch-Schönlein Purpura, lineare IgG Ablagerung bei Patienten mit Goodpasture-Syndrom und eine unregelmäßige IgG und Komplementverteilung bei Patienten mit SLE können diese Erkrankungen ausgeschlossen werden5.
Diagnose
Das American College of Rheumatology hat für die Diagnosestellung des M. Wegener eine Klassifikation herausgebracht, in der mindestens zwei oder mehr Kriterien erfüllt sein müssen4,9:
- pathologisches Urinsediment (Mikrohämaturie, Eryzylinder)
- Entzündliche Nasen- oder Mundschleimhautveränderungen (eitrige oder blutige Rhinorhö, Mundschleimhautulcera)
- Pathologische Thorax-Röntgenaufnahmen (Rundherde, fixierte Infiltrate, Kavernen – einhergehend mit Hämoptoe)
- Nachweis einer granulomatösen Histopathologie (granulomatöse Veränderungen in Gefäßwänden oder peri/extravaskulär.
Diese Merkmale dienen der Unterscheidung anderer Vaskulitiden4.
Differentialdiagnose
- Infektiöse HNO- und Lungenerkrankungen: Bei einer Wegener Granulomatose besteht eine Therapieresistenz gegenüber Antibiotika.10
- Andere Vaskulitiden10: Hier spielen vor allem ANCA-assoziierte Vaskulitiden mit Lungenbefall eine Rolle: z.B.: Churg-Strauss-Syndrom und mikroskopische Polyangiitis7
- Lungenabszess: Bei Kavernenbildung im CT nicht immer einfach abzugrenzen.
- Granuloma gangraenescens: Unspezifische granulomatös-nekrotisierende Entzündung der Nasenschleimhaut. Der WG morphologisch und klinisch sehr ähnlich11.
Therapie
1.Lokal begrenztes Initialstadium:
Monotherapie mit Kortikosteroiden, Azathioprin oder MTX5. Manche Autoren empfehlen auch die Gabe von Cotrimoxazol5. (2×1 Tabl. zu 160mg Trimethroprim und 800mg Sulfamethoxazol10) Längerfristige Remissionen in 2/3 der Fälle10.
2.Generalisationsstadium mit lebens- oder organbedrohlichem Verlauf:
- Remissionsinduktion: Hier werden stärkere Immunsuppressiva zur Krankheitskontrolle verabreicht5
- Firstline Therapie: Gabe von Prednison 1 mg/kg KG/d + CYC 2 mg/kg KG/d oral (max. 200mg) bzw. CYC i.v. (15-20 mg/kg KG)6 als Bolus alle 18 Tage, jeweils mit Uromitexan-Gabe als Blasenschutz5,10. Dabei sollte nach der European League Against Rheumatism (EULAR) die i.v. Bolusgabe von CYC der oralen Gabe vorgezogen werden3. CYC sollte dabei noch bis ein Jahr nach Remission gegeben werden6.
- Bei schwerem Krankheitsverlauf mit Nierenbeteiligung sollten betroffene Patienten nach Möglichkeit immer eine Kombinationstherapie bestehend aus CYC, Kortikosteroiden i.v. und Plasmapherese erhalten5. Reserveoptionen: Antithymozytenglobulin, Infliximab und Rituximab5, das im Rahmen von Studien einen Benefit gezeigt hat3.
- Bei nicht lebensbedrohlichen Organmanifestationen oder leichterem Erkrankungsbild evtl. nur MTX (10-20mg/Woche)8 oder Azathioprin (2mg/kg/KG)8 + Prednison (1mg/kg/KG/d) zur Remissionsinduktion6.
- Erhaltungstherapie: Sie beinhaltet eine weniger aggressive Therapie mit dem Ziel, die Nebenwirkungen zum minimieren unter Beibehaltung der Remission.5
Nach Eintritt einer Remission kann CYC ausgetauscht werden gegen MTX oder Azathioprin5. Bei Nebenwirkungen oder Kontraindikationen gegen Azathioprin Wechsel auf andere Immunsuppresiva wie z.B.: MMF, Cyclosporin oder Leflunomid5. Die Prednisondosis wird in Abhängigkeit der Klinik und Entzündungsparameter langsam reduziert5. 6 Monate nach Therapiebeginn sollte die Dosis 5-10mg/d nicht überschreiten10. Zur Therapie/Prävention einer nasalen Besiedlung mit Staph. aureus wird Cotrimoxazol gegeben, wodurch mehr als 80% der Patienten in Remission bleiben10. Für die Dauer der Erhaltungstherapie gibt es keine gesicherten Empfehlungen10.
Unabhängig vom Stand der Therapie ist es unabdingbar ein Drugmonitoring als auch eine krankheitsspezifische Überwachung durchzuführen, um ein erneutes Aufflammen der Erkrankung, sekundäre Infektionen oder eine mögliche Medikamententoxizität so früh wie möglich zu erkennen5.
Komplikationen
- Massive alveoläre Hämorrhagien einhergehend mit respiratorischer Insuffizienz. Hohe Mortalität2
- Niereninsuffizienz bei ausbleibender Therapie innerhalb 5 Monate bei nachgewiesener Nierenbeteiligung4.
- Entstehung solider Malignome unter immunsuppressiver Therapie5.
Prognose
In der Mehrzahl der Fälle ist die WG eine chronisch rezidivierende Erkrankung, die eine langfristige klinische und Serologische Überwachung erfordert. Während die Nierenbeteiligung ein wesentlicher Faktor für die Frühmorbidität/-mortalität ist, tritt mit zunehmender Krankheitsdauer, die pulmonale Morbidität in den Vordergrund9.
Noch vor 20 Jahren lag die Mortalitätsrate bei M. Wegener bei 80% 1 Jahr nach Diagnosestellung, heute kann mit modernster Therapie eine initiale Remission erzielt werden, obgleich Rückfälle nichts ungewöhnliches darstellen3. Ca. 40-65% der Patienten mit Wegener Granulomatose erleiden trotz Therapie einen Rückfall5,6. Mit Hilfe von Steroiden und Cyclophosphamid kann in bis zu 75% eine komplette Remission erzielt werden4. Je mehr granulomatöse Entzündung sich im Körper findet, desto höher ist die Wahrscheinlichkeit einen Rückfall nach initialer Remission zu bekommen3. Langfristig besteht ein 2,4-faches Risiko eine pulmonale Neubildung zu entwickeln4. Zu beachten sind auch die Nebenwirkungen unter fortlaufender Therapie. Ca. 42% der Patienten im National Institutes of Health series erlitten Folgeerkrankungen aufgrund der CYC –Theapie. Als Beispiel seien Knochenmarkssuppressionen, Infektionen, Unfruchtbarkeit und Blasenerkrankungen genannt.6 Prognostisch ungünstige Faktoren umfassen ein fortgeschrittenes Alter, eine schwere Niereninsuffizienz, eine alveoläre Blutung und eine Positivität für Proteinase(PR)35.
Weiterführende Literatur
Update in the Diagnosis and Management of Pulmonary Vaskulitis
Update in the Diagnosis and Management of Pulmonary Vaskulitis Stephen K. Frankel, Gregory P. Cosgrove, Aryeh Fischer, Richard T. Meehan and Kevin K. Brown Chest 2006;129;452-465Small vessel vasculitis
Small vessel vasculitis Educational Review Paul Brogan & Despina Eleftheriou & Michael Dillon Pediatr Nephrol (2010) 25:1025–1035Histomorphologie interstitieller Lungengerüstveränderungen und pulmonaler Vaskulitiden
Bittmann I, Holl-Ulrich K: Histomorphologie interstitieller Lungengerüstveränderungen und pulmonaler Vaskulitiden. Z Rheumatol 68: 639–649, 2009
Referenzen
1 F. Hua, B. Wilde, S. Dolff, O. Witzke, T-Lymphocytes and Disease Mechanisms in Wegener´s Granulomatosis, Kidney Blood Press Res 2009,32:389-398
2 P. Brogan, D. Eleftheriou, M. Dillon, Small vessel vasculitis, Pediatr Nephrol 2010,25:1025–1035
3 Y. Yazici, M.D., Vasculitis Update, 2007, Bulletin of the NYU Hospital for Joint Diseases 2007;65(3):212-4
4 S. D. Allen, MB BS, MRCS, FRCR and C. J. Harvey, MB BS, MRCP, FRCR, Imaging of Wegener’s granulomatosis, The British Journal of Radiology, 80(2007),757–765
5 S. K. Frankel, G. P. Cosgrove, A. Fischer, R. T. Meehan and K. K. Brown, Update in the Diagnosis and Management of Pulmonary Vasculitis, Chest 2006;129:452-465
6 C. A Langford, Review: Wegener’s granulomatosis: current and upcoming therapies, Arthritis Res Ther 2003, 5:180-191
7 I. Bittmann, K. Holl-Ulrich, Histomorphologie interstitieller Lungengerüstveränderungen und pulmonaler Vaskulitiden, Z Rheumatol 2009,68:639–649
8 H.-P. Hauber, D. Kirsten, Interstitielle Lungenerkrankungen, Z Rheumatol 2009,68:621–629
9 A. Schnabel, W. L. Gross, Pulmonale Vaskulitis-Lungenbeteiligung bei systemischen Gefäßentzündungen, Pneumologie 2000, 54:232-242, Georg Thieme Verlag Stuttgart New York
10 Gerd Herold und Mitarbeiter, Innere Medizin 2010
11 W.Remmele, Pathologie Bd. 3 Leber, Galle, Pankreas, Atemwege, Springer-Verlag Berlin Heidelberg, 2. Auflage 1996
12 C.Thomas, Spezielle Pathologie; Schattauer Stuttgart-New York, 1996
Lehrtexte Spezielle Pathologie

<< Respirationstrakt - Non-Tumor >>
Bilder
Abb. 443: Auf der linken Seite eine mehrkernige Riesenzelle, rechts daneben zwei kleine Nekrosen
Abb. 523: Pulmonale Vaskulitis mit entzündlichen Infiltraten
Respirationstrakt - Non-Tumor - weitere Lehrtexte
Respirationstrakt - Non-Tumor - Kasuistiken
Organpathologie-Atlas
Weiterführende Literatur