Lymphangioleiomyomatose (LAM)
Definition
Die Lymphangioleiomyomatose zählt zu den zystischen Lungenerkrankungen und ist gekennzeichnet durch eine diffuse nicht maligne Proliferation von glatten Muskelfasern in der Wand der Lymphgefäße. Die Lunge ist dabei am häufigsten beteiligt,1 es können aber auch mediastinale, abdominelle,3 und retroperitoneale Lymphknoten betroffen sein.2,4,7 Man unterscheidet eine sporadische Form von einer Form, die im Rahmen der tuberösen Sklerose auftritt.1 Bei ca. ein Drittel der Patienten entwickelt sich eine LAM im Rahmen der tuberösen Sklerose.1,2Epidemiologie
Die LAM ist eine seltene Erkrankung, die ausschließlich Frauen im gebärfähigen Alter betrifft1,2,3,4,5,6,7 und üblicherweise vor dem 50. Lebensjahr in Erscheinung tritt.7 Die Prävalenz wird auf 1:1000000 geschätzt.2,4 Eine erbliche Belastung konnte nicht nachgewiesen werden.7
Pathogenese
Die Ätiologie der LAM ist unklar. Es besteht jedoch ein Zusammenhang mit einer somatischen Mutation des TSC-2-Gens, ein Tumorsuppressorgen, dass mit tuberöser Sklerose assoziiert ist.2,4 Während es sich bei der tuberösen Sklerose um eine Keimbahnmutation handelt, liegt bei der LAM lediglich eine sporadische Mosaikmutation vor. Diese Mutationen betreffen den mTOR-Signaltransduktionsweg. Durch den Ausfall der Inhibition dieses Signalweges kommt es zu einer mTOR-Überexpression, die wiederum die Zellproliferation begünstigt.1
Etwaige Beziehungen zur tuberösen Sklerose werden unter anderem damit diskutiert,5 dass bei einem Drittel der Patientinnen identische Lungenveränderungen wie bei der LAM auftreten.4 Patientinnen mit einer LAM sollten daher sorgfältig auf das Vorliegen einer tuberösen Sklerose gescreent werden.4
Der Umstand, dass die LAM überwiegend bei prämenopausalen Frauen vorkommt und bei einem hohen Östrogen-Rezeptorstatus exazerbiert, macht zwar eine Hormonabhängigkeit wahrscheinlich,3 diese spielt aber nach heutigem Kenntnisstand keine weitere pathogenetische und klinische Rolle mehr, sowie kaum noch eine therapeutische Rolle.4
Makroskopie
Makroskopisch ist die meist vergrößerten2 Lungen von schwammartiger Konsistenz7 und zeigen eine diffuse Durchsetzung mit dünnwandigen Zysten unterschiedlicher Größe. Der Zystendurchmesser variiert dabei von 0,5-2cm, kann aber auch in Einzelfällen bis 10cm betragen und wie bei einem Emphysem vergrößert sein. (Bild der Honigwabenlunge5,2,4 Die befallenen Lymphknoten sind meist verdickt, und haben eine ebenso schwammartige Schnittfläche wie die Lunge.2,5 Der Ductus thoracicus ist erweitert und mit Chylus gefüllt.2,5 Daneben kann auch ein chylöser Aszites bestehen.2,5
Mikroskopie
Histologisch erkennt man diffus, manchmal auch knotig angeordnete ausgeprägte perivasale , peribronchiale und subpleurale Wucherungen von unreifen glatten Muskelfasern (“smooth-muscle-like“-Zellen1 ), die reichlich Lymphspalten einschließen.2,4,7
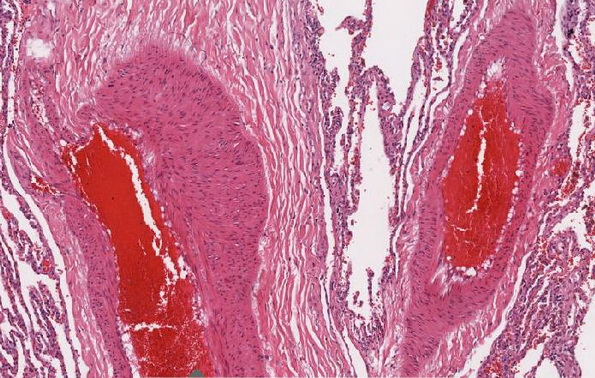
Abb. 478: Perivasale Proliferation glatter Muskelzellen bei einer Patientin mit Lymphangioleiomyomatose
Weiterhin kommt es zu einer Ausbreitung in den Alveolarsepten4 mit papillomatöser Vorwölbung in die Alveolarlichtung.5,7 Diese besonderen glatten Muskelzellen werden heute als LAM-Zellen bezeichnet. Eine Honigwabenlunge tritt in der Regel erst mit fortschreitender Erkrankung auf, und entsteht durch Rupturen im Bereich der Alveolarwände gepaart mit einer Obstruktion der Bronchiolen, die zur Ausbildung herdförmiger zystischer Läsionen führen.4 Ursache dieser Zystenentstehung ist eine Freisetzung von Metalloproteinasen, durch deren proteolytische Aktivität Alveolarsepten zerstört werden, und so letztlich große zystische Läsionen entstehen.4 Wucherungen in den Venenwänden können zur Blutstauung, kapillären Hämorrhagien und zur Siderose führen.2,5 Die Lymphbahnen sind durch obstruierende Muskelwucherungen meist dilatiert, in den Lymphknoten ist das Gewebe durch Muskelwucherungen ersetzt, die lymphgefüllte Hohlraume umgeben.5 Morphologisch lassen sich drei Arten von LAM-Zellen unterscheiden:
1.Spindelförmige Zellen: Sie exprimieren den Proliferationsmarker PCNA, reagieren schwach mit HMB-45 und setzen die Matrix-Metalloproteinase MT1-MMP frei.4
2.Epitheloidzellige Variante: Sie reagiert stark mit HMB-45, ist selten PCNA positiv, und exprimiert nur gering MT1-MMP.4
3.Eine kleine rundliche Form: Diese ist noch wenig charakterisiert.4
Immunhistochemie
Die Muskelzellen zeigen immunhistochemisch eine Positivität für α-Aktin, Desmin, und Vimentin.1,2,4,7 Charakteristisch für LAM Zellen ist weiterhin die Anfärbbarkeit für HMB-45, einem Marker, der das Glykoprotein 100 markiert.1,2,4 Dies ist ein Vorläuferprotein der Eumelaninsynthese, und wird normalerweise in Melanozyten exprimiert. Im Rahmen der LAM-Diagnostik hat es eine Spezifität von 100%.4 Zu beachten gilt allerdings, dass nicht alle LAM Zellen HMB-45 exprimieren, sondern die Expression zwischen 20-70% variieren kann.4 Daneben exprimieren LAM-Zellen noch Hormonrezeptoren. (Östrogen und Progesteron)4
Klinik
Die Patienten klagen häufig über Husten und progrediente Dyspnoe vor allem bei Belastung.2,4,5,7 Ein Pneumothorax wird in 50% der Fälle beobachtet;1,2,4,5,7 ein Emphysem7 oder Hämoptoe5,7 können gelegentlich vorliegen. Selten finden sich ein Chylothorax (10% d. F.),2,4,5,7 und persistierende Thoraxschmerzen.2 Mit dem Fortschreiten der Erkrankung kann sich eine Rechtsherzinsuffizienz entwickeln.5 Gelegentlich können auch trockene Nebengeräusche auskultiert werden.2,4Gastrointestinale Manifestationen mit chylösem Aszites sind dagegen deutlich seltener.3 Weiterhin können vergrößerte retroperitoneale Lymphknoten und Angiomyolipome der Niere abdominelle Beschwerden verursachen.2
Diagnostik
- Lungenfunktion: Erkennbar ist trotz interstitieller Lungenerkrankung meist eine obstruktive (Reduktion der FEV 1 und FEV 1 /FVC2 ) seltener eine gemischt obstruktive/restriktive oder eine rein restriktive Ventilationsstörung.1,2,4 In bis zu 30% der Fälle findet sich eine erhöhte Totalkapazität.2,4 Die CO- Diffusionsmessung ist meist deutlich vermindert und stellt einen der sensitivsten Parameter dar.1,2,4 Die DLCO korreliert direkt mit dem histologischen Progress, und invers mit der Zeit bis zur Transplantation.2 Häufig liegt noch eine Hypoxämie in Ruhe vor.2,3,4
- Bronchioalveoläre Lavage (BAL): Die BAL kann Hinweise auf eine alveoläre Hämorrhagie liefern.4 Daneben können noch zahlreiche eisenpositive hämosiderinhaltige Makrophagen nachgewiesen werden.4 Durch diese Befunde kann die LAM differentialdiagnostisch gut gegenüber der Histiozytose X abgegrenzt werden.4
- Bronchoskopie: Bei Entnahme transbronchialer Biopsien kann die LAM-Diagnose mit Hilfe der Histologie und Immunhistochemie erfolgen.3 Unter Umständen kann eine VATS/Lungenkeil zur Materialgewinnung nötig sein.2,3
- Röntgen Thorax: Die Thoraxaufnahme kann vor allem im Anfangsstadium unauffällig sein.4 Bei einem Drittel der Patienten kommen retikulonoduläre interstitielle Infiltrate und bei 60% zusätzlich Zysten und Bullae zur Darstellung.4
Weiterhin können Zeichen der Überblähung mit tiefstehenden Zwerchfellen und erweiterten Retrosternal- und Retrokardialräumen als auch Kerley-B-Linien und Trübungen beobachtet werden.2,4 - HR-CT: Pathognomonisch für die LAM sind vor allem diffus verteilte multiple dünnwandige Zysten mit einem Durchmesser von 0,5-5 cm.1,2,4 Die Zysten können über die gesamte Lunge verteilt sein, und sind von gesundem Lungengewebe umgeben.1,2,3,4 Je nach Krankheitsstadium können die Zysten in ihrer Form variieren.2 Während zu Beginn der Erkrankung die Zysten klein und rund sind, zeigen sie bei zunehmender Erkrankungsdauer größere Variationen in Form und Größe. (polygonal, ovoid bis hin zu unregelmäßigen Formen)2 Weiterhin kann auch eine Milchglasverschattung bestehen.1,2 Die HR-CT ist die sensitivste Methode für die Diagnose der LAM.4 Im Rahmen der LAM ist das Lungenvolumen meist vergrößert.4
- Abdomensonografie/-CT: Bei einem Teil der Patientinnen werden neben den thorakalen Beschwerden Angiomyolipome der Nieren entdeckt.1,2 Bei ca. 70% finden sich zusätzlich vergrößerte retroperitoneale Lymphknoten. Eine weitere aber seltenere extrapulmonale Manifestation umfasst mit einer Häufigkeit von ca. 20% retroperitoneale Lymphangioleiomyome, welche mit chylöser Flüssigkeit gefüllt sind und u.U. Abdominalschmerzen verursachen können.1,2,3,4
Die Diagnose der LAM wird häufig verzögert gestellt.2,4 In der Regel vergeht ein Zeitraum von bis zu vier Jahren zwischen Beginn der Symptome und der Diagnose.2,4 Die LAM sollte daher bei jeder Patientin mit rezidivierendem Pneumothorax, Emphysem, Chylothorax oder Angiomyolipom differentialdiagnostisch abgeklärt werden.
Differentialdiagnose
Differentialdiagnostisch sollten folgende Erkrankungen in Erwägung gezogen und/oder ausgeschlossen werden:
- Erkrankungen die ebenfalls mit einer glattmuskulären Proliferation einhergehen wie z.B.: die Hyperplasie der glatten Muskulatur im Interstitium bei idiopathischer Lungenfibrose, bei pulmonaler Hypertonie, und beim Emphysem.2,4,7
- Neoplasien der glatten Muskelzellen, wie z.B.: benigne metastasierte Leiomyome,7 Leiomyosarkome und verschiedene zystische Sarkome.2,4
- multiple pulmonale leiomyomatöse Hamartome7
- Histiozytose X2
Therapie
In Anbetracht der Immunhistochemie kann in Einzelfällen ein hormonelles Konzept in Erwägung gezogen werden. (Gabe von Antiöstrogenen und Gestragenen)4
In der aktuellen Literatur findet sich jedoch kein Beleg, dass die Progression der LAM durch diese Therapiemaßnahmen aufgehalten werden kann.1,2 In der neueren Literatur scheint eine Behandlung mit Medroxyprogesteron 800mg i.m. in monatlichen Abständen ein gutes Ansprechen zu zeigen.4 Aus diesem Grund wird ein Therapieversuch mit diesen Medikamenten derzeit empfohlen.1 Entsprechende Studienlagen liegen derzeit jedoch nicht vor und sind abzuwarten.1 Aufgrund der neuen Erkenntnisse zur Pathogenese laufen aktuell Studien zum Einsatz von mTOR-Inhibitoren (Sirolismus).1 Daten aus einer randomisierten placebokontrollierten Studie stehen derzeit noch aus.1 Von einer Ovarektomie sollte nach dem aktuellen Stand der Literatur heute eher Abstand genommen werden.4
In der Regel erfolgt die Therapie jedoch symptomatisch. Die bronchiale Obstruktion kann mit Bronchodilatatoren behandelt werden.3 Ein Pneumothorax wird beim Erstereignis in der Regel mit einer Saugdrainage versorgt, bei Rezidiven kann eine Pleurodese oder eine Pleurektomie in Erwägung gezogen werden.2,3 Beim Vorliegen eines Chylothorax empfiehlt sich zunächst eine fettfreie Diät.3 Bei massiver Ansammlung von Chylus kann auch eine Pleurapunktion erfolgen.2 Nur selten ist die Ligatur des D. thoracicus erforderlich.4 Im Spätstadium bei terminaler pulmonaler Insuffizienz verschafft oft nur eine Lungentransplantation Linderung.2,3,4
Komplikationen
- Bullae und Pneumothorax als Folge der Obstruktion von Bronchiolen durch LAM-Zellproliferate sowie Behinderung des Atemflusses1,4
- Chylothorax und chylöser Aszites durch eine Verengung der Lymphgefäße.1,2,4 In seltenen Fällen kann sich ein chylöser Perikarderguss entwickeln. [2]
- Hämosiderose und Hämoptysen durch Obstruktion von Venolen [2],4,5
- Pulmonale Hypertonie mit Cor pulmonale und respiratorischem Versagen2
Prognose
Nach früheren Studien war eine progrediente respiratorische Insuffizienz6 einhergehend mit dem Tod der Patienten innerhalb von 10 Jahren nach Diagnosestellung assoziiert.2,7 Nach neueren Untersuchungen ist die Prognose, u.a. bedingt durch eine frühere Diagnosestellung, wesentlich besser mit einer aktuellen 10-Jahres Überlebensrate von etwa 80% und einem 15-Jahresüberleben von etwa 70%.4 Ein Stillstand der Erkrankung ist trotz häufiger Progredienz möglich.4 Bei erfolgter Lungentransplantation besteht eine 5-Jahres-Überlebensrate von 51%.4 Über ein erneutes Auftreten einer LAM im Transplantat wurde vereinzelt berichtet.1,3,4 Die Prognose von LAM-Patientinnen nach Lungentransplantation ist besser als von anderen Patientengruppen mit Lungen-Tx.1
Weiterführende Literatur
Seltene zystische Lungenerkrankungen: Lymphangioleiomyomatose, pulmonale Langerhanszell-Histiozytose, lymphozytäre interstitielle Pneumonie
Prasse A, Kayser G. Seltene zystische Lungenerkrankungen: Lymphangioleiomyomatose, pulmonale Langerhanszell-Histiozytose, lymphozytäre interstitielle Pneumonie. Deutsche Medizinische Wochenschrift. 2010 Mar;135(10):461-5. Epub 2010 Mar 2.From the Archives of the AFIP Lymphangioleiomyomatosis: Radiologic-Pathologic Correlation
G.F. Abbott, MD et al.; From the Archives of the AFIP Lymphangioleiomyomatosis: Radiologic-Pathologic Correlation; RadioGraphics 2005; 25:803-828Lymphangioleiomyomatose (LAM): seltene Ursache von Aszites und Pleuraergüssen
A. R. J. Schneider et al., Lymphangioleiomyomatose (LAM): seltene Ursache von Aszites und Pleuraergüssen, Dtsch Med Wochenschr 2004; 129: 1375–1378,Georg Thieme Verlag Stuttgart-New YorkLymphangioleiomyomatose
Seltene Lungenerkrankungen: Pulmonale Lymphangioleiomyomatose U. Costabel, J. Guzman; Pulmonale Lymphangioleiomyomatose; Pneumologie 2002; 56:309-315
Referenzen
1 A. Prasse, G. Kayser; Seltene zystische Lungenerkrankungen: Lymphangioleiomyomatose, pulmonale Langerhanszell-Histiozytose, lymphozytäre interstitielle Pneumonie, Dtsch Med Wochenschr 2010; 135: 461-465, Georg Thieme Verlag Stuttgart-New York
2 G.F. Abbott, MD et al.; From the Archives of the AFIP Lymphangioleiomyomatosis: Radiologic-Pathologic Correlation; RadioGraphics 2005; 25:803-828
3 A. R. J. Schneider et al., Lymphangioleiomyomatose (LAM): seltene Ursache von Aszites und Pleuraergüssen, Dtsch Med Wochenschr 2004; 129: 1375–1378,Georg Thieme Verlag Stuttgart-New York
4 U. Costabel, J. Guzman, Pulmonale Lymphangioleiomyomatose; Pneumologie 2002; 56: 309-315, Georg Thieme Verlage Stuttgart-New York
5 W.Remmele, Pathologie Bd. 2 Verdauungstrakt, Springer-Verlag Berlin Heidelberg, 2. Auflage 1996
6 W. Böcker, Pathologie; Elsevier GmbH, München, 4. Auflage 2008
7 C.Thomas, Spezielle Pathologie; Schattauer Stuttgart-New York, 1996
Lehrtexte Spezielle Pathologie

<< Respirationstrakt - Non-Tumor >>
Bilder
Abb. 478: Perivasale Proliferation glatter Muskelzellen bei einer Patientin mit Lymphangioleiomyomatose
Respirationstrakt - Non-Tumor - weitere Lehrtexte
Respirationstrakt - Non-Tumor - Kasuistiken
Organpathologie-Atlas
Weiterführende Literatur