Schilddrüsenkarzinome
Definition
Bei den Tumoren der Schilddrüse unterscheidet man differenzierte Karzinome (papilläres (PTC) und follikuläres (FTC ) Schilddrüsenkarzinom), undifferenzierte Karzinome (anaplastisches Karzinom (ATC)), gering differenzierte Karzinome (PDTC), medulläre Karzinome (MTC), Plattenepithelkarzinome und seltenere Varianten wie Sarkome, Hämangiosarkome, oder Metastasen anderer Tumoren wie z.B.: RCC, Mamma-CA, maligne Melanome und BC48.
- Die follikulären Karzinome (FTC ) der Schilddrüse umfassen alle maligne epitheliale Tumoren mit follikulärer Differenzierung ohne histologische (Papillen) und/oder zytologische Merkmale (Milchglaskerne) des papillären Karzinoms11011.
- Das papilläre Karzinom (PTC ) ist ein von den Follikelzellen ausgehender maligner Tumor mit papillären und/oder follikulären Strukturen sowie charakteristischen Kernveränderungen (Milchglaskerne, dachziegelartige Überlappung, Kernkerben)14. (WHO-Definition)
- Die WHO-Klassifikation hat zu den 4 Entitäten noch das gering differenzierte Schilddrüsenkarzinom (PDTC ) eingeführt, welches eine biologische und morphologische Position zwischen den differenzierten Karzinomen ( FTC und PTC )und dem ATC einnimmt1347. Es macht mit deutlichen geographischen Unterschieden 4-7% der Karzinome mit Follikelzellursprung aus4 und entwickelt sich überwiegend aus differenzierten Karzinomen14.
- Die anaplastischen Karzinome (ATC ) sind undifferenzierte, hochmaligne Tumoren mit vermutetem Follikelzellursprung, bei denen definitionsgemäß immunhistochemisch kein Thyreoglobulin mehr nachweisbar ist14.
- Das medulläre Karzinom (MTC ) ist ein maligner Tumor mit phänotypischer C-Zell-Differenzierung10. Es kommt zur Produktion von Kalzitonin, calcitonin gene peptide, sowie CEA, weniger häufig von Somatostatin, GRP, Serotonin, ACTH oder Prostaglandinen10. Beim neuroendokrinen Phänotyp der Tumorzellen ist Synaptophysin und Chromogranin A nachweisbar10. Weiterhin kommt es häufig zur Einlagerung von Amyloid im Stroma10.
Epidemiologie
Schilddrüsenkarzinome sind mit einer jährlichen Erkrankungsrate von 4,1/100000 bei Frauen und 1,5/100000 bei Männern eine seltene Tumorerkrankung1. Deutschland gibt es jährlich ca. 2500 Neuerkrankungen8. Die Inzidenz steigt mit dem Alter11. Der Altersgipfel liegt bei den differenzierten Karzinomen um das 50. Lebensjahr (beim papillären Karzinom gibt es auch einen Gipfel um das 25. Lebensjahr) und bei den enddifferenzierten Karzinomen um das 60. Lebensjahr8. Frauen sind 2,5mal häufiger betroffen als Männer8. Die malignen Tumoren machen bei der Frau ca. 1,5% beim Mann ca. 0,5% aller malignen Tumoren aus1. Von solitären Tumorknoten sind 80-90% Adenome und 10-20% Karzinome10. In Strumen treten Tumoren häufiger auf, als in der nicht vergrößerten Schilddrüse10.Die überwiegende Anzahl der Karzinome der Schilddrüse nehmen ihren Ursprung von den Follikelzellen. (88-99% aller Schilddrüsenkarzinome)
- Das FTC ist mit 10-30%4 der häufigste maligne Schilddrüsentumor im Kropfendemiegebiet1011. Die Inzidenz sinkt mit dem Rückgang der Stumainzidenz bei Jodprophylaxe10. Frauen sind hier bis zu 5mal häufiger betroffen als Männer14.
- Das PTC ist mit >80% der häufigste maligne SD-Tumor in ausreichend jodversorgenden Gegenden1. Im Strumaendemiegebiet ist es mit 30-40% etwa gleich häufig wie das Follikuläre Karzinom1. Es tritt bevorzugt bei jüngeren Patienten auf (80% unter 40 Jahren)111.
- Das ATC macht derzeit 2-15% der malignen Schilddrüsenkarzinome aus1. Es tritt meist im Alter von über 60 Jahren auf10 und kommt nur in 5% der Fälle vor dem 50. Lebensjahr vor111. Die Inzidenz ist im Endemiegebiet erhöht.11 Die Tumoren entwickeln sich meist aus einem lange bestehenden Knotenkropf14.
- Der Anteil der MTC beträgt mit deutlichen regionalen Schwankungen 1-12%111. Er kann sporadisch (80%) oder familiär (autosomal dominanter Erbgang11 )gehäuft auftreten10. 15-20% der medullären Schilddrüsenkarzinome sind mit einer MEN 2a oder b kombiniert10. Der Tumor tritt dann bereits bei Kindern10, bzw. zwischen dem 20. Und 30 Lebensjahr auf147. Die nicht hereditären sporadischen Formen finden sich hingegen überwiegend bei über 45-jährigen Patienten14.
Ätiologie und Pathogenese
Bei der Entstehung von SD-Tumoren spielen epigenetische (Jodmangel, Strahlung, Rauchen) und genetische Faktoren (weibliches Geschlecht, familiäre Prädisposition) eine wesentliche Rolle34.
Die der Versorgung mit Jod in Kropfgebieten ist assoziiert mit fallender Inzidenz follikulärer Karzinome und steigender Inzidenz papillären Karzinome7.
Externe Strahlung spielt bei der Entwicklung von papillären Tumoren vermutlich eine Rolle7. Die durchschnittliche Zeit zwischen Strahlenbelastung und Tumorentwicklung beträgt ca. 20 Jahre, kann aber variieren7.
Papilläre Karzinome wurden bei Patienten mit FAP, Cowden-Syndrom, HNPCC und Peutz-Jeghers-Syndrom beschrieben7. FAP wird durch die Keimbahnmutationen des APC-Gen verursacht7. Schilddrüsenkarzinome, meist papilläre Karzinome (mehr als 95% der Fälle), treten in 1% bis 2% der Patienten mit FAP auf7. Alle diese Patienten weisen Keimbahnmutationen des APC-Gens, jedoch keine somatischen Mutationen oder Verlust der Heterozygotie (LOH) für das APC-Gen in Schilddrüsentumoren auf7. Interessanterweise zeigen die meisten dieser Tumoren eine Aktivierung von RET/ptc1 in Schilddrüsentumoren, was auf einen möglichen Zusammenhang zwischen APC und RET / PTC in der Entwicklung dieser speziellen Untergruppe der familiären papillären Karzinom stehen könnte7.
Molekularpathologie
Nur ein geringer Teil der SD-Karzinome entsteht durch Keimbahnmutationen im Rahmen von definierten, i.d.R. autosomal dominant vererbten Syndromen (MEN 2, FAP)34. Die sporadischen Tumoren der SD sind monoklonale Neoplasien, die durch Proliferation einer durch eine somatische Mutation genetisch modifizierten Vorläuferzelle entstanden sind34. Eine oxidative Schädigung der DNA durch endogene reaktive Sauerstoffspezies gilt als ein wesentlicher Mechanismus der Mutationsentstehung34.
- Bei ca. 80% aller PTC können charakteristische genetische Veränderungen nachgewiesen werden, es hat aber keine definierte Vorläuferläsion34. Etwa 50% der Tumoren zeigen eine somatische Punktmutation des BRAF-Gens10 (überwiegend ältere Patienten), etwa 10-20% weisen eine RET/PTC-Rearrangement10 (überwiegend Kinder betroffen) und etwa 10% ein NTRK1-Rearrangement auf34. Die genannten Rearrangements sind überwiegend in Zusammenhang mit strahleninduzierten Tumoren beobachtet worden34.
- Beim FTC sind RAS-Mutationen7, PAX8-Rearrangements710 und eine Deregulation des Transkriptionsfaktors FOXO зa nachweisbar34. Weiterhin konnten Mutationen und Amplifikationen des die PIз K kodierenden PIзKCA-Gens, inaktivierende Mutationen im PTEN-Gen mit nachfolgender AKT-Aktivierung als auch die Überexpression von AKT selbst zur konstitutiven Aktivierung der PIзK/AKT-Kaskade nachgewiesen werden34.
- Beim PDTC wurden bisher keine eigenständigen Charakteristika gefunden34. Am häufigsten werden Mutationen des RAS-Gens und des p53 Tumorsuppressorgens gefunden, während BRAF-Mutationen eher selten sind34.
- ATC entstehen entweder aus vorbestehenden differenzierten oder gering differenzierten Schilddrüsenkarzinomen oder de novo34. Das ATC stellt einen genetischen Zustand der maximalen Signaltransduktionsaktivierung dar, als dessen Auslöser die Inaktivierung des p 53-Tumorsuppressorgens angesehen wird34. Es besteht eine kumulative Aktivierung multipler Tyrosinkinase-Kaskaden, wobei nach derzeitigem Kenntnisstand dabei dem PI 3K/AKT-Signalweg eine Schlüsselrolle zukommt34.
- Etwa 25% der MTC sind genetisch determiniert und treten autosomal dominant vererbt im Rahmen eines MEN 2 Syndroms auf, familiäre MTC entwickeln sich aus einer „neoplastischen C-Zell Hyperplasie“4. Der Genlokus für die MEN und das isolierte familiäre medulläre Schilddrüsenkarzinom befindet sich auf Chromosom 10q11.2, hier wurden auch die ersten verantwortlichen Keimbahnmutationen des RET-Protoonkogens identifiziert234.
Makroskopie und Mikroskopie
| Follikuläre Karzinom – minimalinvasive Variante | |
|---|---|
| Makroskopie | Die Tumoren machen 37-50%134 der follikulären Karzinome aus. Sie werden von einer breiten, dicken bindegewebigen Kapsel begrenzt7. Die Schnittfläche imponiert teils grau-weiß, bei oxyphilen Karzinomen auch gelb-braun; häufig finden sich Einblutungen und hyaline regressive Veränderungen11. |
| Mikroskopie | Der Tumor zeigt ein mikrofollikuläres bis trabekuläres Wachstumsmuster mit regulären, schmalen runden Follikeln7. Einblutungen und Nekrosen können vorkommen, eine signifikante mitotische Aktivität findet sich häufig7. Wichtige Kriterien für die Abgrenzung zum Adenom: 1. Einzelne Einbrüche in Gefäße innerhalb oder jenseits der Tumorkapsel (Tumorthrombus im Gefäß), 2. Kapseldurchbruch (Kontakt zwischen Tumorepithel und umgebenden nicht-neoplastischem Schilddrüsenparenchym).13411 Der Nachweis multipler Gefäßeinbrüche oder Kapseldurchbrüche schließt die Diagnose eines minimal invasiven Karzinoms aus11, der Tumor ist hier bereits als grob-invasiv einzustufen1. |
| Follikuläre Karzinom – grobinvasive Variante | |
|---|---|
| Makroskopie | Die Tumoren haben eine grau-weiße Schnittfläche mit unscharfer Begrenzung111. |
| Mikroskopie | Bei diesen Tumoren findet sich ein hoch differenziertes follikelbildendes bis solid-trabekuläres Strukturmuster, eine geringe Tumorzellpleomorphie und wenig Mitosen1. Der Nachweis multipler Gefäßeinbrüche oder Kapseldurchbrüche ist wegweisend für die Diagnose Grob-invasives Karzinom3411. |
- Varianten: Onkozytäre Tumoren haben eine schlechtere Prognose als nichtonkozytäre follikuläre Karzinome, bedingt durch ihre geringere Radiojodspeicherfähigkeit4. Die Abgrenzung der hellzelligen Variante des follikulären Karzinoms gegenüber einer Metastase eines RCC gelingt bereits im HE-Schnitt, kann aber auch durch den Nachweis der Thyreoglobulinsynthese erfolgen411.
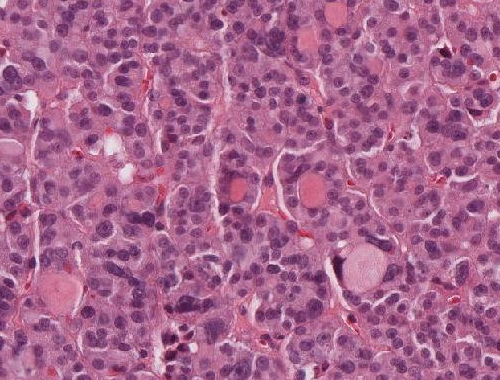
Abb. 533: Follikuläres Schilddrüsenkarzinom; erkennbar ist ein follikelbildendes Strukturmuster mit regulären, schmalen runden Follikeln eine geringe Tumorzellpleomorphie und wenig Mitosen
| Papillares Karzinom | |
|---|---|
| Makroskopie | In der Mehrzahl der Fälle findet sich überall in der Schilddrüse lokalisiert7 makroskopisch ein fester, solitärer nicht umkapselter Tumor mit grau-weißer Schnittfläche1711. Es kann jedoch auch eine Kapsel vorhanden sein10. Mehr als 80% der papillären Karzinome liegen intrathyreoidal1, 10-20% überschreiten die SD-Kapsel111. Per Definitionem sind typische papilläre Karzinome größer als 1 bis 1.5 cm; oft messen sie zwischen 2 und 3 cm7. Eine läsionale Verkalkung ist möglich7. Aufgrund umfangreicher Sklerosierung kann die Läsion einer Narbe ähneln7. |
| Mikroskopie | Die papillären Karzinome können in wechselndem Ausmaß sowohl follikuläre als auch papilläre Anteile enthalten711. Die papillären Strukturen bestehen aus fibrovaskularen, manchmal hyalin-verquollenen baumartigen Verästelungen, die von kubischen bis zylindrischen Zellen umkleidet werden11. Fallweise finden sich im Stroma histiozytäre Schaumzellen11. Follikuläre Anteile bestehen aus unreifen trabekulären Formationen oder aus hochdifferenzierten neoplastischen Follikeln11. Charakteristisch sind helle, milchglasartige und teilweise dachziegelartig übereinanderliegende Kerne711, eine unregelmäßig geformte Kernmembran (Rosinenkern)10, und Kernkerbungen – „nuclear grooving”7. Zudem können Zytoplasmaeinschlüsse im Kern auftreten (Kerneinschlüsse)10; Nukleoli sind wenn vorhanden klein, selten finden sich Mitosen11. Psammomkörperchen sind in etwa 40% bis 50% der Fälle vorhanden, ihre Präsenz in Schilddrüsengewebe zeigt, dass sich wahrscheinlich ein papilläres Karzinom irgendwo in der vorliegenden Drüse befindent7. Der Befund Psammomkörperchen in einem zervikalen Lymphknoten ist ein starker Beweis für ein papillares Kazinom in der Schilddrüse7. |
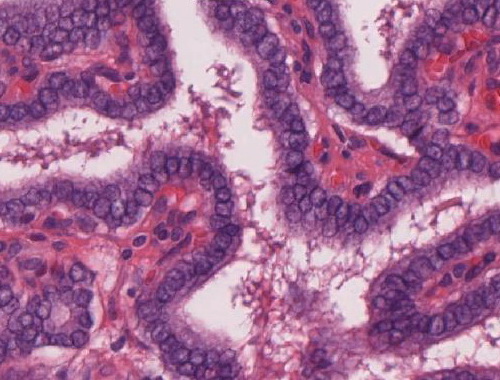
Abb. 534: Papilläres Schilddrüsenkarzinom, erkennbar sind helle milchglasartige und teilweise dachziegelartig übereinanderliegende Kerne.
- Besonderheiten: In der WHO-Klassifikation werden neben einen konventionellen Typ 15 Varianten des PTC aufgeführt4, die aufgrund ihrer klinischen Relevanz im Befundbericht vermerkt werden sollten: 1. Papilläres Mirkokarzinom (<1cm7 ), 2. Makrofollikuläre Variante, 3. Follikuläre Variante, 4. Diffus sklerosierendes Karzinom, 5. Onkozytäre Variante, 6. Hochprismatische („tall cell“) Variante und 7. Kolumnäre Variante, 8. Klarzellige Variante, 9. Solide Variante, 10 Kribriforme Variante, 11 Variante mit fasziitisähnlichem Stroma, Mischdifferenzierte Varianten: 12. PTC mit fokaler insulärer Komponente, 13. PTC mit Plattenepithel- oder Mukoepidermoidkarzinom, 14, PTC mit Spindel- oder Riesenzellkarzinom, 15. Kombiniertes papilläres und medulläres Karzinom4. In etwa 25% der papillären Karzinome besteht eine Assoziation mit einer diffusen lymphozytären Thyreoiditis11. Die diffus sklerosierunde Variante des papillären Karzinoms ist hingegen mit einer Autoimmunthyreoiditis assoziiert11. 10% der papillären Karzinome finden sich in einer Knotenstruma11|
| Gering differenziertes Karzinom | |
|---|---|
| Makroskopie | Erkennbar sind große7, auffällig scharf begrenzte grau-weiße Tumoren mit angedeutet lobulärer Schnittfläche1 und ausgedehnten Nekrosen7. |
| Mikroskopie | Mikroskopisch besteht der Tumor aus unterschiedlich großen Zellnestern und -verbänden1 und zeigt ein neuroendokrines Wachstumsmuster7. Daneben finden sich in unterschiedlichem Ausmaß follikuläre und/oder papilläre Strukturmuster1. Die histologische Diagnose eines PDTC („Turin-Proposal“) beruht auf dem Nachweis eines soliden, trabekulären und/oder insulären Aufbaus, sowie von mindestens einem der folgenden Kriterien: Nekrosen, eine Mitoserate von >3/10 HPF und/oder spezifische Kernveränderungen („convoluted nuclei“)34. |
| Immunhistologie | Die Tumoren zeigen eine deutlich verminderte Thyreoglobulin-Expression bei verringerter oder meist normaler nukleärer Expression von TTF-1 auf34. |
| Anaplastische Karzinom | |
|---|---|
| Makroskopie | Makroskopisch ist eine ausgedehnte tumoröse Durchsetzung der Schilddrüse typisch1711. Sehr selten kann sich auch ein kleines Karzinom als anaplastisch herausstellen11. Der graue Tumor ist von weicher Konsistenz10. |
| Mikroskopie | Mikroskopisch findet man überwiegend Tumoren mit pleomorphen, spindeligen7 oder epitheloiden Zellen, die einzelne oder mehrere teilweise bizarre Kerne enthalten4, massenhaft atypische Mitosen4 sowie meist ausgedehnte Nekrosen7 und Einblutungen111. Daneben finden sich Infiltrate durch neutrophile Granulozyten4. Die Tumorzellen zeigen manchmal eine deutliche Dissoziation, was den Eindruck eines Sarkoms hervorruft111. In kompakteren Tumorabschnitten überwiegen spindelige Zellen111. In seltenen Fällen liegt Osteoid oder Knorpel vor111. Ein Teil dieser Tumoren ist durch den Reichtum an osteoklastären Riesenzellen charakterisiert14711. Der Tumor bricht in Venen (mit Obliteration des Lumens4 ) ein und durchwandert oft die Organkapsel und wächst in die umgebenden Strukturen10. |
| Immunhistochemie | Immunhistochemisch exprimieren anaplastische Karzinome kein Thyreoglobulin11. Nachweisbares Thyreoglobulin stammt entweder aus präexistenten nicht-neoplastischen Follikeln oder deutet auf einen differenzierten Anteil eines anaplastischen Karzinoms hin11. Zytokeratin ist nur in 60%, Vimentin praktisch in allen Fällen darstellbar111. Beim immunhistochemischen Nachweis von epithelialen Anteilen (Zytokeratin) sollte auch bei gleichzeitig bestehender Vimentin-Positivität die Diagnose eines anaplastischen Karzinoms gestellt werden11 |
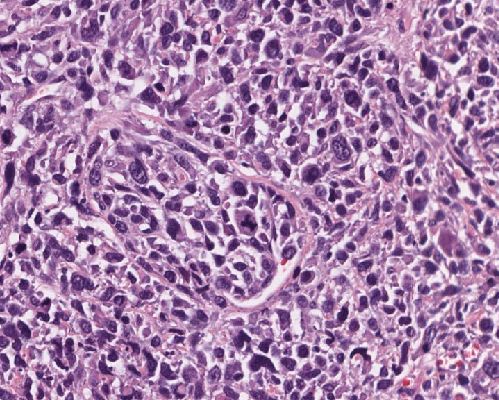
Abb. 532: Undifferenziertes Tumorgewebe mit ausgeprägter Kernpleomorphie.
| Medulläres Karzinom | |
|---|---|
| Makroskopie | Bei der sporadischen Form findet sich ein scharf begrenzter, manchmal auch bekapselter grau-weißer Tumor1 mit fallweise bestehenden granulären Verkalkungen auf der Schnittfläche11. Bei der familiären Form liegt dagegen ein bilaterales multifokales Wachstum7 mit unterschiedlich großen, überwiegend unscharf begrenzten grau-weißen Herden vor111. Die Tumoren finden sich in der Regel im Bereich der höchsten C-Zell-Konzentration, also in den seitlichen oberen zwei Drittel der Drüse7. Die Größe der Tumoren kann von kaum sichtbar bis mehrere Zentimeter variieren7. |
| Mikroskopie | Histologisch kann das Bild stark variieren. Der Tumor kann solide, follikuläre, oder seltener (pseudo-) papilläre oder kleinzellige Areale enthalten10. Die Zellen sind rundoval oder spindelig, und in Platten angeordnet, das Stroma enthält oft Amyloid (60-80%1411 ), in dem u.a. ProCalcitonin nachgewiesen werden kann710. Der Amyloidnachweis ist jedoch nicht wegweisen für die Diagnose7. Mitosen können vorkommen7. Bis zu 40% der Tumoren enthalten extrazellulär Muzin, intrazelluläres Muzin findet sich hingegen nur in 15% der Zellen7. |
| Immunhistochemie | Bei kombinierter Anwendung von Calcitonin und Chromogranin A-Antikörpern werden praktisch alle medullären Karzinome erfasst; CEA findet sich in der Mehrzahl der Karzinome, wobei insbesondere spindelzellige und mäßig differenzierte Varianten stärker positiv sind1411. |
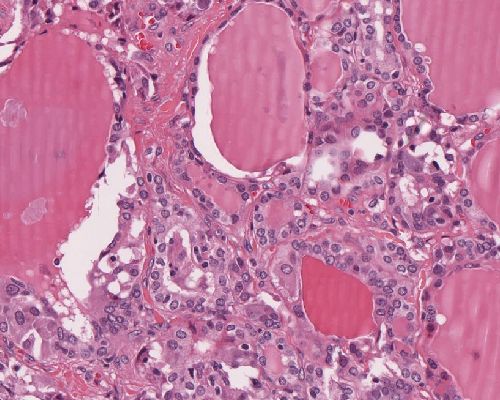
Abb. 579: Medulläres Schilddrüsenkarzinom mit follikulären und soliden Differenzierungsmustern
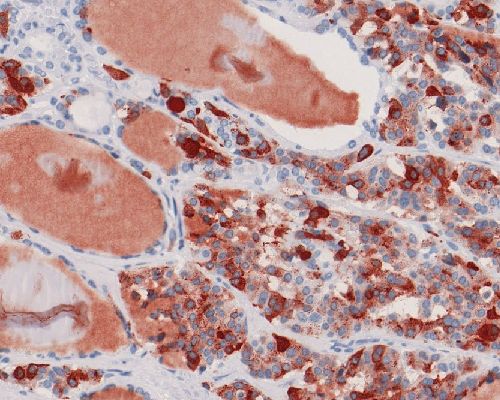
Abb. 580: Stark positive Immunreaktion der Tumorzellen für Calcitonin.
Immunhistochemie
Jeder ungewöhnliche Tumor der Schilddrüse sollte immunhistochemisch untersucht werden. Folgende Antikörper sind in der Schilddrüsendiagnostik hilfreich1:
- Thyreoglobulin: Es dient dem Nachweis des Follikelzellursprungs eines Schilddrüsentumors15.
- Calcitonin: Dieser Marker dient dem Nachweis eines medullären Karzinoms5, sowie einer C-Zell-Hyperplasie1. Calcitonin ist auch in etwa 50% der kleinzellig-anaplastischer Bronchialkarzinome sowie fokal in hyperplastischen Nebenschilddrüsen nachweisbar1.
- CEA: Es ist üblicherweise in medullären Karzinomen positiv; insbesondere bei gering differenzierten Karzinomen ist CEA dem Calcitonin als Marker überlegen1.
- Chromogranin A: Dies ist ein neuroendokriner Marker15. Es ist positiv in medullären Karzinomen, Metastasen neuroendokriner/kleinzellig-anaplastischer Karzinome der Schilddrüse, Paragangliomen der Schilddrüse, sowie in normalen, hyperplastischen und neoplastischen Nebenschilddrüsen1.
- TTF -1: Dieser Marker kann hilfreich beim Nachweis des Schiddrüsenursprungs eines Tumors gegenüber Metastasen sein1. Es ist ein nukleär positiver Marker in normalen, hyperplastischen und neoplastischem Schiddrüsengewebe1. Medulläre Karzinome können auch TTF-1 negativ sein1, anaplastische Schilddrüsenkarzinome sind immer negativ5 Achtung: u.a. sind kleinzellig-anaplastische und Adenokarzinome der Lunge für TTF-1 positiv15.
- Endothelzellmarker (CD 34, Faktor VIII): Sie dienen dem Nachweis eines malignen Hämangioendothelioms der Schilddrüse1.
- CK 14: Dieser Marker dient einer oxyphilen Differenzierung5.
- Zytokeratin: Nachweis der epithelialen Natur eines Tumors. In anaplastischen Karzinomen gelingt der Nachweis nur in etwa 60% der Fälle1.
- LCA : Marker der B- bzw. T-Zellreihe (CD3 bzw. CD20), CD 5, CD10 Antikörper gegen die Immunglobulinleichtketten Kappa bzw. Lamba5. Dieser Marker sollte bei allen kleinzellig-anaplastischen Tumoren der Schilddrüse zum Nachweis/Ausschluss eines malignen Lymphoms eingesetzt werden1.
- Parathormon: Nachweis ektopen Schilddrüsengewebes15.
- Galectin-3: Ein in den meisten papillären und dem überwiegenden Teil der follikulären Schilddrüsenkarzinomen exprimiertes Lektin1. Es kommt aber auch fallweise in pseudopapillären Hyperplasien und Adenomen vor und kann auch teilweise in medullären Karzinomen nachgewiesen werden1.
Differentialdiagnose
- Metastasen anderer Tumoren (Nierenzellkarzinom, Lunge, Mamma, GI-Trakt, maliges Melanom)1
- Fibrosierende Prozesse der Schilddrüse (subakute Thyreoiditis de Quervain, Hashimoto Thyreoiditis)1
- Riedelstruma18
- Angiosarkom der Schilddrüse1
- Fibro- oder Leiomyosarkom der Schilddrüse1
- Schwellung der Halslymphknoten (Entzündungen, Mononukleose, HIV-Lymphadenopathie, Metastasen, maligne Lymphome)8
Klinik
Hauptsymptom ist meist ein indolenter tastbarer Strumaknoten von derber, harter9 Konsistenz8. Bei den anaplastischen Karzinomen kann ein schnelles Wachstum richtungsweisend sein8. Ein langsameres Wachstum über Wochen bis Monate findet man hingegen bei den differenzierten Karzinomen8. Daneben finden sich in der Regel vergrößerte zervikale8 und supraklavikuläre9 Lymphknoten, die noch vor dem Primärtumor sichtbar werden können8.
Lokale Spätsymptome/Komplikationen umfassen eine derbe fixierte höckrige9 Struma (schlechte Verschieblichkeit), LK-Schwellung (zervikal und/oder supraklavikulär9, Heiserkeit (Recurrensparese), Atemnot und Stridor, Hornersyndrom, Schluckbeschwerden, Hals-, Ohren-, und Hinterhauptschmerzen9 sowie eine obere Einflussstauung8.
Diagnostik
Die lokale Diagnostik umfasst:
- Anamnese und klinische Untersuchung: Hier sollte nach einer stattgehabten Radiatio im Halsbereich (vor 10-20 Jahren)8 oder anderer Strahlenexposition, nach einer MEN in der Familienanamnese und nach einem Strumawachstum trotz ausreichender Substitution gefragt werden9. # Klinische Untersuchung: Sie umfasst die Palpation der Schilddrüse (derb palpabler Knoten, Verwachsungen mit der Haut) und den zervikalen Lymphknotenstatus8.
- Labor: Thyreoglobulin und CEA als Tumormarker für follikuläres und papilläres Karzinom (nur für die Verlaufskontrolle in der Nachsorge geeignet)8. Beim Verdacht auf ein medulläres Schilddrüsenkarzinom sollte das basale Calcitonin bestimmt werden2. Ist dieser Wert erhöht, kann die Diagnose eines medullären Karzinoms gestellt werden, und ein Stimulationstest mit Pentagastrin ist nicht notwendig2. Nur bei grenzwertig erhöhten Werten kann dieser zur Diagnosesicherung notwendig werden2. Neben der Diagnostik ist das Calcitonin der entscheidende Parameter in der Postoperativen Verlaufskontrolle2. Bei allen Patienten sollte weiterhin präoperativ das Serumkalzium bestimmt werden, um einen begleitenden Hyperparathyreoidismus als Hinweis auf ein MEN-Syndrom auszuschließen2.
- Sonografie der Schilddrüse: Zu beurteilen sind die Echostruktur (solide vs. zystisch), die Beziehung zu Nachbarstrukuren und Verkalkungen8. Unregelmäßig begrenzte echoarme Areale können verdächtig auf ein Malignom sein9. Zusätzlich können pathologisch vergrößerte Lymphknoten erkannt werden2.
- Schilddrüsenszinitgrafie: Ein kalter Knoten ist in 1-5% der Fälle ein Karzinom8. Dies gilt für Deutschland, da hier ein Endemiegebiet vorliegt. In Gebieten mit niedriger Strumarate, z.B.: bei Trinkwasseriodierung ist die Karzinomwahrscheinlichkeit eines kalten Knoten viel höher (bis zu 30%)8.
- OP: Bei fortbestehendem Malignomverdacht, auch bei negativer Zytologie, hat eine OP mit histologischer Aufarbeitung zu erfolgen9. Intraoperativ hat die Gefrierschnittdiagnostik mit Auswirkung auf die Operationsstrategie und –radikalität einen hohen Stellenwert1.
- Genanalyse: Beim C-Zellkarzinom sollte eine Genanalyse auf eine Punktmutation im RET-Protoonkogen erfolgen, sowie eine genetische Beratung und Familienuntersuchung angeboten werden9.
- Punktionszytologie/FNA wichtige Entscheidungshilfe zur Operationsindikation und/oder Verifizierung eines klinischen Malignitätsverdachtes1. Die Sensitivität der Methode liegt bei 68-98% und die Spezifität bei 72-100%5. Die Rate falsch negativer Befunde wird in der Literatur mit 1,5-11,5%, die Rate falsch positiver zytologischer Befunde mit 0-8% angegeben5. Die FNA erlaubt mit hoher Treffsicherheit die Diagnose eines papillären, medullären, gering differenzierten und anaplastischen Schilddrüsenkarzinoms5. Bei Tumoren mit follikulärem Aufbau ist die Methode jedoch limitiert (zytologisch kann ein Gefäßeinbruch und/oder Kapseldurchbruch nicht dargestellt werden)5. Zytologische Beurteilung des Schilddrüsenpunktats5
- Hintergrund: Beschaffenheit des Kolloid, Siderophagen, Lymphozyten, Riesenzellen, Blut
- Zellanordnung: mikro- vs. Makrofollikulär, papillär
- Zellzahl: inadäquat, repräsentativ, vermehrt
- Morphologie der Zellen: Zellgröße, uniform oder variabel, Kerneinschlüsse, Inzisuren, Nukleoli, oxyphile Differenzierung
| Zytologie5 | |
|---|---|
| Normalbefund | Isomorphe follikuläre Strukturen, zweidimensionale Zellverbände, sowie einzeln gelegene nackte Kerne von Thyreozyten |
| Papilläres Karzinom | Dreidimensionale papilläre Strukturen |
| Medulläres- und gering differenziertes Karzinom | größere und kleinere Zellverbände mit deutlich unregelmäßig gelagerten Kernen neben dissoziierten Zellen |
| anaplastisches Karzinom | hochgradig atypische, meist einzeln gelegene Tumorzellen neben Zelldetritus und gelegentlich auch Entzündungszellen |
| Papilläres Karzinom | Zylindrische und polygonale Zellen |
| Medulläres Karzinom | trianguläre und spindelige Zellen mit manchmal bereits erkennbarem granulären Zytoplasma |
Die Umfelddiagnostik umfasst:
- Sonografie des Abdomens: Detektion von Lebermetastasen, im Zweifel CT-Abdomen nötig; bei Verdacht auf ein Phäochromozytom im Rahmen eines MEN ist eine Sonografie der Nebennieren nötig2.
- Röntgen: CT/MRT-Hals und CT-Thorax9; zur Darstellung können Einwachsungen in umliegendes Gewebe, LK-Metastasen und Fernmetastasen in der Lunge kommen. (CAVE: Bei follikulären und papillären Karzinomen darf kein iodhaltiges Kontrastmittel verwendet werden!8)
- Ganzkörperszintigrafie des Skelettsystems: Sie dient der Darstellung von Metastasen8. Als Tracer können radioakiv markierte Thyreoglobulin-AK kommen8. # PET: Generell: Lokalisation persistierender und rezidivierender Schilddrüsenkarzinome, sowie gleichzeitiger Ausschluss bzw. Nachweis von Fernmetastasen6. Zu beachten ist, dass nicht bei allen Fällen Rezidive zur Darstellung kommen6. Die PET-CT eignet sich besonders für den Nachweis oxyphiler und radiojodnegativer differenzierter Schilddrüsenkarzinomrezidive6. Somatostatinrezeptor-Szintigrafie bei medullären Karzinomen zur Detektion von Leber- oder Lungenmetastasen; in der Detektion von kleinen Tumorrezidiven im zervikalen Bereich allerdings nur eine Sensitivität von 33%2.
Therapie
- Jedes nachgewiesene Karzinom bzw. jeder suspekte Befund sollte chirurgisch saniert und histologisch aufgearbeitet werden8. In der Regel erfolgt eine totale Thyreoidektomie (inkl. Kapsel) mit zervikozentraler Lymphonodektomie8.
- Bei PTC >1cm besteht die Indikation zur totalen Thyreoidektomie, bei nicht organkapselinfiltrierenden solitären PTC <1cm ohne Metastasen ist eine totale Thyroidektomie hingegen nicht erforderlich6. Treten diese Karzinome jedoch multifokal auf, liegt eine Organkapselinvasion oder ein schlechter Differenzierungsgrad vor, wird die radikale Primärtherapie empfohlen6. Die therapeutische Lymphknotendissektion ist bei diesen Tumoren internationaler Bestandteil aktuell gültiger Behandlungsempfehlungen6.
- Bei breit invasiven FTC hat aufgrund des Fernmetastasenrisikos in jedem Fall eine totale Thyreoidektomie mit postoperativer Radiojodablation zu erfolgen6. Bei der minimalinvasiven Variante ist bei fehlender Angioinvasion unabhängig von der Tumorgröße hingegen eine primäre Lobektomie ohne weiterführende Therapie ausreichend67. Eine Indikation zur prophylaktischen Lymphknotendissektion besteht hingegen nicht6.
- Bei resektablen PDTC und ATC ist die Indikation zur Thyreoidektomie in aller Regel gegeben und sollte auch bei Organüberschreitung angestrebt werden6. Liegt beim ATC bereits eine Infiltration umliegender Strukturen vor, kann eine palliative Operation zur Reduktion der Tumormasse erfolgen8.
Eine befallsorientierte Lymphknotendissektion ist anzustreben6. - Bei den MTC ist die primäre Thyreoidektomie in allen Stadien und bei allen Formen gegeben6. Bei Genträgern einer zum hereditären MTC disponierenden RET-Mutation ist der beste Zeitpunkt für eine prophylaktische Thyreoidektomie wenn unabhängig von Alter ein basal noch nicht erhöhtes Calcitonin vorliegt, da bei dieser Konstellation noch keine LK-Metastasen nachgewiesen werden konnten6. Weiterhin hat sich eine kompartmentorientierte Lymphknotendissektion durchgesetzt6.
- Bei Fehlen einer progredienten Fernmetastasierung vor allem bei differenzierten Karzinomen und bei MTC ist bei Rezidiven grundsätzlich die Indikation zur erneuten Resektion gegeben6. Hinzu kommt das Rezidive im Zervikal-und Mediastinalbereich beträchtliche tumorbedingte Komplikationsrisiken aufweisen und diesen mit einem Zweiteingriff vorgebeugt werden sollen6.
- Postoperativ sollte bei den differenzierten Schilddrüsenkarzinomen nach ca. 10-14 Tagen eine adjuvante Radio-Iod-Therapie erfolgen8. Zu Beginn wird ein Jod-131 Ganzkörperscan zur Tumordarstellung und Metastasensuche angefertigt8. In mehreren Fraktionen wird dann hochdosiertes Jod-131 verabreicht, bis kein iodspeicherndes Gewebe szintigrafisch mehr nachgewiesen werden kann8. Der postoperative TSH-Anstieg aufgrund des Fehlens der SD-Hormone ist dabei von Vorteil8. Das ATC und das MTC nehmen nicht am Iodumsatz teil und sind daher für eine adjuvante Radioiodtherapie nicht zugänglich8.
- Die Indikationen zur externen Strahlentherapie beschränken sich heute auf die additive Therapiesituation bei Irresektabilität6. Beim ATC ist die externe Radiatio des Tumors und der Metastasen fester Bestandteil des multinodalen Therapiekonzepts6, da dieser Tumor strahlenempfindlicher als andere SD-Karzinome ist9. Da MTC strahlenresistent sind, ist die radikale Operation und die radikale en-bloc Lymphadenektomie2 prognoseentscheidend9.
- Alternative ist eine palliative Chemotherapie im Rahmen von Studien bei inoperablen, nicht radioiodspeichernden Schilddrüsenkarzinomen, und rasch progredienten MTC9.
- Nach erfolgreicher Radioiodtherapie folgt eine lebenslange hochdosierte T4-Substitution (Euthyrox ®, 150-200mg), um die TSH-Produktion so niedrig wie möglich zu halten8. TSH-Zielbereich: <0,1 mU/l9 → verminderter Reiz auf evtl. noch vorhandene Metastasen →Rezidivprophylaxe8.
Prognose
- Das FTC metastasiert charakteristischer Weise hämatogen in die Lungen, das Skelettsystem und in das Gehirn191011 und die Leber7. Fernmetastasen treten bei rund 10% der minimalinvasiven4 und 80% der grob-invasiven Karzinome auf1. Lymphknotenmetastasen sind selterner147 und treten erst ab einer Tumorgröße von ca. 2cm auf4. Metastasen bekapselter Tumoren können auch noch bis zu 5 Jahren nach der initialen Resektion auftreten7. Die Prognose bei minimal invasiven Karzinomen ist mit einer 10-Jahres-Überlebensrate von >90-95% sehr günstig11011. Hier spielt die primäre Operationsradikalität eine wichtige Rolle11. Bei den grob-invasiven Karzinomen beträgt die 10-Jahres-Überlebensrate nur 30-50%1. Als prognostisch ungünstige Faktoren gelten7:
- Präsenz von Metastasen
- Alter >50 Jahre
- große Tumoren
- ausgeprägte Gefäßinvasion
- kapselüberschreitendes Wachstum
- schlecht differenzierte Anteile innerhalb des Tumors
- Die durchschnittliche 10-Jahres-Überlebensrate des PTC liegt bei 80-90%1, wobei papilläre Mikrokarzinome eine praktisch 100%-tige Langzeit-Überlebensrate zeigen1411. Gefäßeinbrüche in Venen mit hämatogener Metastasierung sind mit 5-7%7 selten und erfolgt, wenn überhaupt erst sehr spät, dann vor allem in die Lunge10. Die lymphogene Tumorausbreitung ist hingegen die Regel11. Initiale LK-Metastasen (Lunge und Knochen7 ) finden sich in Abhängigkeit vom Tumortyp in 25-84% der Fälle11 und treten klinisch oft schon vor dem eigentlichen Primärtumor auf, der in vielen Fällen eine minimale Größe aufweist, aber dennoch multizentrisch vorhanden sein kann (unauffälliges Szintigramm der SD)9. Als prognostisch ungünstige Faktoren gelten11:
- Die 5 Jahres Überlebensrate beim PDTC beträgt zwischen 30-65%, die 10-Jahres- Überlebensrate 25-35%4. Initiale Metastasen finden sich häufig in der Lunge und im Skelettsystem2. Es konnte gezeigt werden, dass das Ausmaß der schlecht differenzierten Komponente in einem gut differenzierten Schilddrüsen-Tumor die Prognose beeinflussen kann; Tumoren mit mehr als 10% der schlecht differenzierten Komponente gehen häufiger mit Rezidiven, regionalen Metastasen, Fernmetastasen und einer schlechten Prognose einher7.
- Das ATC entwickelt sich üblicherweise in einer länger bestehenden Knotenstruma mit plötzlicher Vergrößerung sowie Schluck- und/oder Atembeschwerden11. In vielen Fällen liegt zum Zeitpunkt der Diagnose bereits ein Einwachsen in umliegende Strukturen wie Karotiden, Larynx, Trachea oder Ösophagus vor11. Das ATC hat eine außergewöhnlich schlechte Prognose11. Der Tumor ist hochmaligne und metastasiert früh lymphogen und hämatogen10. Trotz Therapie versterben die meisten Patienten innerhalb eines Jahres an den Folgen des lokalen Tumorwachstums1, oder an Fernmetastasen11. Bevorzugte Metastasierungsorte umfassen Lungen, Nebennieren, Skelett, Gehirn und Herz11. Die 2-Jahres-Überlebensrate liegt unter 10%, die 5-Jahres-Überlebensrate unter 5%111. Von etwas günstigerer prognostischer Bedeutung scheint der Nachweis differenzierter Anteile am ATC zu sein111.
- Das MTC metastasiert meist früh lymphogen in die regionären Halslymphknoten, später hämatogen in Lunge, Leber10 und Knochen7. In bis zu 50% der Fälle finden sich Lymphknotenmetastasen zum Diagnosezeitpunkt, in etwa 15% bis 25% der Fälle.Fernmetastasen7. Das spontan auftretende MTC zeigt beim Fehlen von LK-Metastasen zum Zeitpunkt der Diagnosestellung eine gute Prognose (Heilungsraten zwischen 80-100%2 ); die mittlere 10-Jahres-Überlebensrate bei Vorhandensein von LK-Metastasen beträgt dagegen nur 40%10. Als schlechte prognostische Faktoren gelten11:
Genetisch determinierte MTC im Rahmen einer MEN 2 treten wesentlich früher im Leben auf (meist schon vor dem 30. Lebensjahr11 ) und verlaufen teilweise aggressiver10. Aufgrund der frühzeitigen chirurgischen Intervention bei betroffenen Mitgliedern aus Familien mit MTC beträgt die 5-Jahres Überlebensrate zwischenzeitlich 94%, die 10-Jahres Rate 85%1.
Weiterführende Literatur
Tumoren der Schilddrüse
Schmid KW, Sheu SY, Görges R, Ensinger C, Tötsch M. Tumoren der Schilddrüse. Pathologe. 24: 357-72, 2003Medulläres Schilddrüsenkarzinom
Mann B, Kasten C, Hotz H, Buhr HJ. Medulläres Schilddrüsenkarzinom. Onkologe. 2000. 6:651–659.Molekularpathologie von Schilddrüsentumoren
Schmid KW. Molekularpathologie von Schilddrüsentumoren. Pathologe: DOI 10.1007/s00292-010-1321-2, 2010Klassifikation von Schilddrüsenkarzinomen
Schmid KW: Pathogenese, Klassifikation und Histologie von Schilddrüsenkarzinomen. 16: 644–656, 2010Präoperative Punktionszytologie beim Schilddrüsenkarzinom
Tötsch M, Quadbeck B, Görges R, Schmid KW. Präoperative Punktionszytologie beim Schilddrüsenkarzinom. Onkologe. 2005. 11:40–49.Molekulare Pathogenese von Schilddrüsentumoren
Karger S, Krause K, Führer D. Molekulare Pathogenese von Schilddrüsentumoren. Deutsche Medizinische Wochenschrift. 2006 Jul 28;131(30):1671-4.
Referenzen
1 K. W. Schmid, S.-Y. Sheu, R. Görges, C. Ensinger, M. Tötsch, Tumoren der Schilddrüse, Pathologe 2003, 24:357-372
2 B. Mann, c. Kasten, H. Hotz, H.J. Buhr, Medulläres Schilddrüsenkarzinom, Onkologe 2000, 6:651-659
3 K.W.Schmid, Molekularpathologie von Schilddrüsentumoren, Pathologe 2010, [Suppl2] 31:[afp]-[alp]
4 K.W.Schmid, Pathogenese, Klassifikation und Histologie von Schilddrüsenkarzinomen, Onkologe 2010, 16:644-656
5 M. Tötsch, B. Quadbeck, R. Görges, K.W.Schmid, Präoperative Punktionszytologie beim Schilddrüsenkarzinom, Onkologe 2005, 11:40-49
6 H. Dralle, K. Lorenz, A.Machens, M. Brauckhoff, P. Nguyen Thanh, Tumortyp- und tumorstadienorientiertes chirurgisches Konzept bei Karzinomen der Schilddrüse, Onkologe 2010, 16:666-677
7 S. E. Mills, D. Carter, J. K. Greenson, und V. E. Reuter, Sternberg’s Diagnostic Surgical Pathology, Lippincott Williams & Wilkins, Auflage: 4th revised edition
8 M. Müller, Chirurgie für Studium und Praxis 2010/11: Unter Berücksichtigung des Gegenstandskataloges und der mündlichen Examina in den Ärztlichen Prüfungen
9 Gerd Herold und Mitarbeiter, Innere Medizin 2010
10 W. Böcker, Pathologie; Elsevier GmbH, München, 4. Auflage 2008
11 W. Remmele, Pathologie Bd. 3 Leber, Galle, Pankreas, Atemwege, Springer-Verlag Berlin Heidelberg, 2. Auflage 1996
Lehrtexte Spezielle Pathologie

Schilddrüse - weitere Lehrtexte
Schilddrüse - Kasuistiken
Organpathologie-Atlas
Weiterführende Literatur
- Schilddrüse (12)