5.6 Gewebsblagerungen
Hyalin Extrazelluläre Ablagerungen einer homogenen, stark eosinophilen Substanz. Dabei handelt sich meist um heterogene Eiweißsubstanzen; z.B. um eine hyaline Degeneration kollagener Fasern.
Amyloid
Homogene extrazelluläre Eiweißablagerungen mit β-Faltblatt-Struktur, am häufigsten handelt es sich um Immunglobulinen (leichte Ketten).
5.6.1 Hyalin
Als Hyalin bezeichnet man extrazelluläre Ablagerungen einer homogenen und stark eosinophilen Substanz. Das Hyalin kann Amyloid ähneln; es handelt sich hierbei jedoch um heterogene Eiweißsubstanzen (z.B. um eine hyaline Degeneration kollagener Fasern). Man unterscheidet verschiedene Arten von Hyalin:
- Bindegewebiges Hyalin z.B.: degeneriertes Kollagen
- Hyaline Plaques: z.B.: im Peritoneum, in der Pleura oder in der Milzkapsel (“Zuckergußmilz”)
- Hämatogenes Hyalin z.B.: Plasmaproteine, alte Makrothromben, hyaline Mikrothromben oder “Kautschukhyalin” (altes Blut in Endometriosezysten oder Strumen)
- Vaskuläres Hyalin z.B.: Plasmaproteine, die sich an Wänden kleiner Gefäße (Arteriolen und Kapillaren) oder in der Niere (Arteriolosklerose, diffuse noduläre Sklerose) ablagern.
Hyaline Ablagerungen haben meist keinen Krankheitswert (Bsp. Corpus luteum / Corpus albicans, hyalinisierte Glomeruli, silikotische Schwielen).
5.6.2 Amyloid
Als Amyloid bezeichnet man eine Gruppe von Proteinen und Glykoproteinen, die zu Gewebeablagerungen führen und folgende Eigenschaften besitzen:
- β-Faltblattstruktur
- charakteristische färberische Eigenschaften (grünliche Polarisation bei Kongorot-Färbung, sog. Dichroismus)
- extrazelluläre Ablagerungen, oft auf Basalmembranen
- resistent gegenüber natürlicher Degradation
- resultierender Verlust der Elastizität des Gewebes mit steifer, wächserner, und manchmal auch speckiger Schnittfläche
Die β-Faltblattstruktur ist wichtig, da der Organismus keine Enzyme hat, die große Moleküle mit β-Faltblattstruktur abbauen können, so dass es zu dauerhaften Gewebsablagerungen kommt. Am häufigsten handelt es sich um abgelagerte Bruchstücke von Immunglobulinen (leichte Ketten) in fibrillärer Form. Man unterscheidet nach der Ätiologie zwei Hauptformen der Amyloidose:
- Primäre Amyloidose: Diese Form der Amyloidose wird als primär bezeichnet, da eine Grundkrankheit fehlt. Es finden sich Ablagerungen in einzelnen Organen (z.B.: Altersamyloid am Herzen oder ein Altersdiabetes der Langerhansschen Inseln).
- Sekundäre Amyloidose: Hier handelt es sich um eine Begleitamyloidose bei anderen Grundkrankheiten. Dabei kommt es in der Regel zu perikapillären Ablagerungen in verschiedenen Organen (generalisierte Amyloidose). Als Grunderkrankungen kommen in Frage:
- Chronische Entzündungen, z.B. Tuberkulose, Colitis ulcerosa, rheumatoide Arthritis und chronische Pyelonephritis (Ablagerung von AA-Amyloid)
- Immunglobulin-bildende Tumoren des lymphatischen Systems (z.B.: Plasmozytom und M. Waldenström (Ablagerung von AL-Amyloid)).
Diagnostisch kann zum Nachweis einer generalisierten Amyloidose eine Haut-, eine Fettgewebs- oder eine tiefe Rektumbiopsie durchgeführt werden. Bei der primären Amyloidose werden die betroffene Organe biopsiert. Die Amyloidose verursacht funktionelle Störungen, die je nach Organbeteiligung verschieden ausfallen können. Die Amyloidnephrose ist charakteristisch beim Befall der Nieren. Betroffen sind dabei vor allem Glomeruli und kleine Gefäße. Es kann zu einer chronischen Niereninsuffizienz kommen. Im Herzen bewirken die interstitiellen Amyloidablagerungen eine verminderte Elastizität, mit der Folge einer einer chronischen Herzinsuffizienz (restriktive Kardiomyopathie).
Die Veränderungen in Leber und Milz und die Amyloidablagerungen in anderen Organen führen seltener zu Krankheitserscheinungen.
Klassifikation der Amyloidose (Beispiele)
| Erkrankung | Amyloid-Typ | Vorstufe |
|---|---|---|
| Systemische Amyloidose | ||
| Multiples Myelom | AL | Ig-Lambda (oder Kappa) Ketten |
| Chronische Entzündungen, Rheumatoide Arthritis, Tuberkulose | AA | SAA |
| Malignome, M. Hodgkin | AA | SAA |
| langjährige Hämodialyse | Aß2M | ß2-Microglobulin |
| Heredofamiliäre Amyloidose, Familiäres Mittelmeerfieber, Familiäres Amyloid, Polyneuropathie | verschiedene | verschiedene |
| Lokalisierte Amyloidose | ||
| Senile kardiale Amyloidose | ATTR | Transthyretin |
| Senile zerebrale Amyloidose: M. Alzheimer | Amyloid-β Amyloid | Precursor Protein (APP) |
| Endokrine Tumoren, Medulläres Karzinom der Schilddrüse | AF | Calcitonin |
Skript Allgemeinpathologie
Bilder (4)
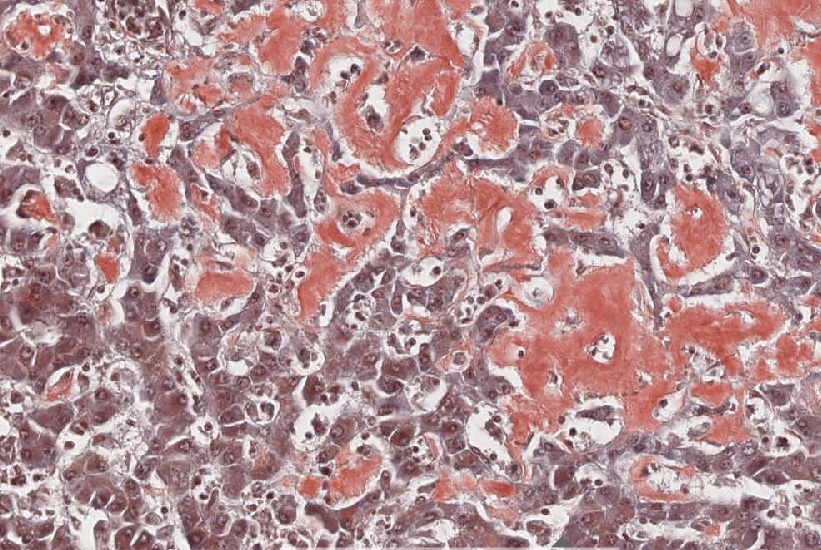
Abb. 31: Amyloidose der Leber: Ablagerung eosinophilen, hyalinen Materials in den Gefäßwänden und Sinusoiden. Atrophie der Hepatozyten.
5 Stoffwechselstörungen - Weitere Themen
5.3 Gewebsblagerungen - Kurspräparate
Selbstevaluation (1)
Bilder (1)