64 j. Patientin mit Proteinurie
Aktueller Befund
Die Patientin wird zur Abklärung einer Proteinurie stationär ins Nierenzentrum Heidelberg aufgenommen.
Körperlicher Untersuchungsbefund
64-jährige schlanke Patientin in guten AZ, RR bei Aufname 120/60 mmHg, HF 100/min, Temperatur 36,6°C, Gewicht bei Aufnahme 81 kg. Integument unauffällig.
EKG
SR, HF 75/min, ID-Typ, keine signifikanten Erregungsrückbildungsstörungen
Anamnese
6 Jahre vor der Nierenbiopsie wurde erstmalig eine nephrotische Proteinurie festgestellt. Zwei Jahre vor der PE sei das Kreatinin bereits auf 1,6 mg/dl angestiegen, und 18 Monate vor der PE wurde es mit 2,2 mg/dl dokumentiert.
Procedere
Nierenbiopsie zum Ausschluß einer glomerulären Erkrankung
Virtuelle Mikroskopie
HE-Färbung
Diagnose
Primäre membranöse Glomerulopathie
Lichtmikroskopie
Die Lichtmikroskopie (HE oder PAS) zeigt eine diffuse, gleichmäßige Verdickung der glomerulären Basalmembran. Die Basalmembran nimmt das Aussehen eines dicken eosinophilen Bandes an. Mit diesen Färbungen kann man aber die Ursache der Basalmembranverdickung nicht erkennen (d.h. die Unterscheidung zwischen einer Verdickung aufgrund der Ablagerung von Immunkomplexen oder einer gesteigerten Produktion der Basalmembranmatrix ist nicht möglich). Diese Veränderungen können jedoch in einer Silber-Methenamin Färbung oder elektronenmikroskopisch dargestellt werden. Mit diesen Techniken wird die Basalmembranmatrix angefärbt, jedoch nicht die Immunkomplexe. Mit dieser Färbetechnik wurden charakteristische ultrastrukturelle Veränderungen in der Basalmembran von Ehrenreich und Churg beschrieben. Die erste Phase ist die Bildung von subepithelialen Immunkomplexen, danach folgt die Synthese neuer Basalmembransubstanzen, die sie einschließen und schließlich werden die Immunkomplexe abgebaut und sind nicht mehr nachweisbar.
Klassifikation der membranösen Glomerulopathie nach Ehrenreich und Churg:
| Stadium | Elektronenmikroskopischer Befund |
|---|---|
| I | Subepitheliale elektronendichte Ablagerungen |
| II | Subepitheliale elektronendichte Ablagerungen mit dazwischenliegenden Basalmembran (Spikes) |
| III | Einbeziehung der subepithelialen elektronendichte Ablagerungen in der Basalmembran |
| IV | Reabsorption der Ablagerungen mit Verlust der elektronendichten Ablagerungen und Aufhellung der Basalmembran. |
Klinik und Differentialdiagnose
Leitsymptom Proteinurie
Bei einer Proteinurie ist die Albuminurie von einer Bence-Jones-Proteinurie zu unterscheiden. Die erfolgt durch Proteinelektrophorese. Von einer Mikroalbuminurie spricht man bei einem Wert von >30–300 mg Albumin. Sie zeigt bei Diabetikern (insbesondere Typ 1) den Beginn einer diabetischen Nephropathie an. Bei Werten von >300 mg/24 h liegt eine Makroalbuminurie vor. Von einer Proteinurie spricht man bei >3g/24 h Gesamtprotien im Sammelurin. Eine Albuminurie bzw. eine Proteinurie kann durch Nierenerkrankung, aber auch durch starke körperliche Belastung, Herzinsuffizienz, Fieber oder Orthostase hervorgerufen werden. Sammelt der Patient getrennt während der Liegezeit (Nachtzeit), so kann eine harmlose “orthostatische” Proteinurie erfasst werden. Für den Patienten auffällig ist das Schäumen des Urins.
Die einfachste Möglichkeit des Nachweises in der Praxis ist ein Streifentest (z.B. Combur). Fällt dieser für Eiweiß positiv aus, sollte die Bestimmung der Gesamtproteinurie aus dem 24-h-Urin erfolgen.
Proteinurien können nach verschiedenen Gesichtspunkten unterteilt werden: klinisch interessant neben der pro Tag ausgeschiedene Menge, vor allem auch das Spektrum der Urinproteine, das einen Hinweis auf den Ursprung der Proteinurie liefern kann. Zur Differenzierung von Proteinen im Urin dienen elektrophoretische Untersuchungen im Urin. Dabei ist zum Vergleich immer auch eine Elektrophorese im Serum durchzuführen. Die Protein-Elektrophorese im Urin wird heute meistens als Agarose-Gel-Elektrophorese (AGE) durchgeführt. Allgemein spricht die Ausscheidung von Albumin und makromolekularen Proteinen (Transferrin, Immunglobuline usw.) für eine glomeruläre Schädigung. Man unterscheidet:
- Selektive Proteinurie: die anionische Ladung der Basalmembran kann bei glomerulären Erkrankungen durch Auflagerungen von Immunglobulinen, Komplementfaktoren oder anderen Molekülen mehr oder weniger neutralisiert werden. Das hat zur Folge, dass bei erhaltener Molekülgrössen-Selektivität, die Selektion der verschiedenen Proteine bezüglich elektrischer Ladung verloren geht. Diese Form der Proteinurie wird daher als selektive, glomeruläre Proteinurie bezeichnet und typischerweise bei Glomerulonephritiden und diabetischer Proteinurie in der Frühphase gesehen. Bei der Urinuntersuchung äussert sie sich in einer vermehrten Ausscheidung von Albumin.
- unselektiv-glomeruläre Proteinurie: Mit zunehmender Schädigung der Basalmembran nimmt auch die Grössenselektion ab, so dass zusätzlich Proteine mit einem Molekulargewicht von mehr als 100’000 im Urin auftreten. Dies betrifft insbesondere Immunglobuline. Man spricht von Bence-Jones
- mikromolekulare (tubuläre) Proteinurie: Bei der mikromolekularen bzw. tubulären Proteinurie führt eine verminderte Rückresorption der kleinmolekularen Proteine zu einer vermehrten Ausscheidung insbesondere von a1- a2- und ß2- Mikroglobulin sowie Lysozym.
Im vorliegenden Fall liegt eine “große Proteinurie” vor. Zusätzlich hat die Patientin Ödeme, Hyperlipidämie, Gerinnungsstörung, nephrotisches Sediment, aber keine Niereninsuffizienz. Dieser Symtomenkomplex wird als nephrotisches Syndrom bezeichnet.
Pathogenese und Klinik des neprotischen Syndroms
Beim nephrotischen Syndrom handelt es sich allgemein um Folgesymptome einer Proteinurie. und ist in der Regel ein Hinweis für eine primär renale Erkrankung oder eine Grunderkrankung mit sekundärer renaler Beteiligung (z. B. Diabetes mellitus und zahlreiche andere Erkrankungen). Der Diabetes mellitus ist die häufigste Ursache eines sekundären nephrotischen Syndroms!
Wenn der renale Eiweißverlust die Synthesekapazität der Leber übersteigt, entwickelt sich der volle Symptomenkomplex des nephrotischen Syndroms mit Ödemen, Hyperlipoproteinämie, Hyperkoagulabilität, Abwehrschwäche. Ursächlich für die zum Eiweißverlust in den Urin führenden Schädigungen sind Ablagerungen von Immunkomplexen oder Amyloid im Bereich des glomerulären Filters sowie dessen Schädigung durch Zytokine.
Die nephrotisches Syndrom ist charakterisiert durch folgende klinische Symptome
- massive Proteinurie, mit den täglichen Verlust Protein im Urin von 3,5 gm oder mehr bei Erwachsenen,
- Hypoalbuminemie, mit Plasma-Albumin von weniger als 3 g / dl;
- generalisierte Ödeme, die offensichtlichste klinische Manifestation, und
- Hyperlipidämie und Lipidurie. Zu Beginn kaum oder gar keine Azotemie, Hämaturie oder Hypertonie.
Die Symptome des nephrotischen Syndrom stehen mit einer kausalen Beziehung zueinander. Das intiale Ereignis ist eine Verdickung der glomerulären kapillären Membranen, die zu erhöhten Durchlässigkeit für Plasmaproteine führt. Es wird daran erinnert, dass die glomeruläre Kapillarwand mit seinen Endothel, GBM, und Podozyten, eine Barrierefunktion besitzt, durch die das glomeruläre Filtrat muss. Eine erhöhten Durchlässigkeit resultiert entweder aus strukturellen oder physikalisch-chemischen Veränderungen. Das generalisierte Ödem beim nephrotischen Syndrom ist, ist eine Folge der Senkung des Plasma-Kolloid-osmotischen Druckes als Folge der Hypoalbuminemie. Die Entstehung der Hyperlipidämie ist unklar.
Die relativen Häufigkeiten der verschiedenen Ursachen der nephrotisches Syndroms variiert je nach Alter. Bei Kindern zwischen 1 und 7 Jahren, ist das nephrotisches Syndrom ist fast immer durch primäre Nierenerkranung verursacht. Bei Erwachsenen dagegen meist eine renale Manifestation einer systemischen Erkrankung. Die häufigste systemische Ursachen der nephrotisches Syndrom bei Erwachsenen sind Diabetes, Amyloidose, und SLE. Die wichtigsten glomerulären Schädigungen beim nephrotischen Syndrom sind die segmentale Glomerulosklerose und die minimal-change-Krankheit (MCD). Letzteres ist wichtig bei Kindern, die erstgenannte Veränderung bei Erwachsenen. Zwei weitere primäre Glomerulopathien, die membranöse Nephropathie und membranoproliferative GN, verursachen ebenfalls das nephrotisches Syndrom. Diese vier Glomerulopathien können histologisch voneinander unterschieden werden.
Indikation zur Nierenbiopsie:
Die Indikation zur Nierenbiopsie beim nephrotischen Syndrom wird vom Alter des Patienten abhängig gemacht und davon ob eine Systemerkrankung vorliegt. Bei Kindern gibt man zunächst hochdosiert Corticosteroide da hier meist eine Minimal-Change-Glomerulopathie vorliegt. Nur bei atypischer Symptomatik oder Nichtansprechen auf Corticosteroide wird bei Kindern beim nephrotischen Syndrom initial eine Nierenbiopsie durchgeführt.
Bei Erwachsenen mit nephrotischem Syndrom ist die häufigste Ursache eine sekundäre Beteilgung der Niere bei Systemerkrankugen, z.B. eine Amyloidose. Zum Ausschluss primärer glomerulärer Erkrankungen ( idiopathische membranöse Glomerulonephritis, fokale segmentale Glomerulosklerose, membranoproliferative Glomerulonephritis, IgA Nephropathie) führt man bei Erwachsenen initial eine Nierenbiopsie durch, um die medikamentöse Therapie festlegen zu können.
Kasuistiken

Bilder

Abb. 16: Glomeruläre Veränderungen
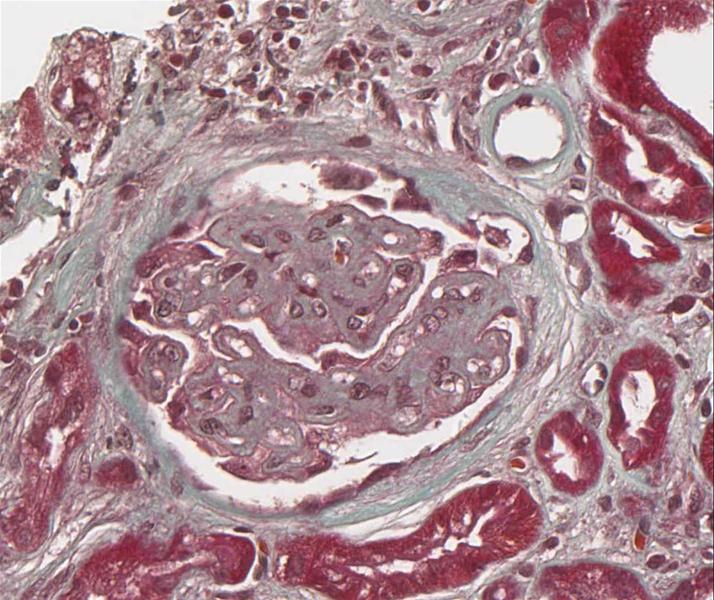
Abb. 17: Glomeruläre Veränderungen
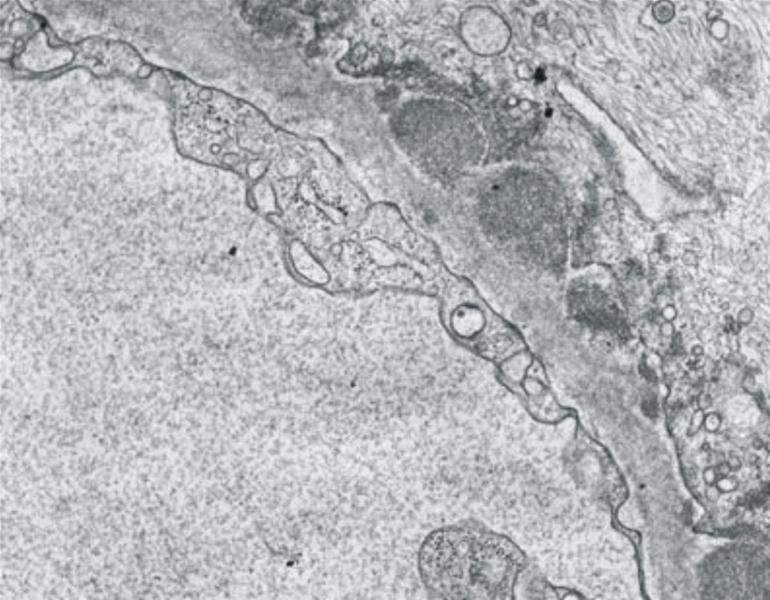
Abb. 18: Elektronenmikroskopische Veränderungen der glomerulären Basalmembran
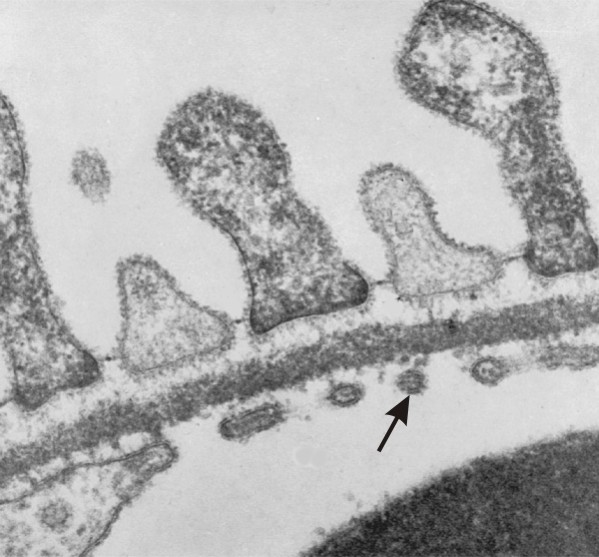
Abb. 19: Normale Verhältnisse der glomerulären Basalmembran (zum Vergleich mit den pathologieschen Veränderungen).
Nephropathologie - weitere Kasuistiken
Nephropathologie - Literatur
Organpathologie-Atlas