Familiäre adenomatöse Polyposis (FAP)
Definition
Die familiäre adenomatöse Polyposis (FAP) ist eine seltene, autosomal-dominant vererbte Erkrankung, die durch das Auftreten einer großen Anzahl von kolorektalen Adenomen (von 100 bis über 1000) charakterisiert ist.
Allgemeines
Bei der klassischen familiären adenomatösen Polyposis (FAP) sind bei 95% der Patienten bis zum 35. Lebensjahr Polypen im Kolorektum klinisch nachweisbar; daneben gibt es aber auch attenuierte Verläufe11. Das Lebenszeitrisiko ein kolorektales Karzinom zu entwickeln beträgt bei ausbleibender Therapie 100%4. Der Nachweis eines kolorektalen Karzinoms erfolgt für gewöhnlich zwischen dem 40. und 50. Lebensjahr, wenn die Erkrankung nicht vorher diagnostiziert und therapiert wurde1789. Bei ca. 7% der Patienten sind die Tumoren schon im Alter von 21, bei 95% jedoch erst mit dem 50. Lebensjahr nachweisbar2. Nicht desto trotz entstehen weniger als 0,5% aller kolorektalen Karzinome auf dem Boden einer FAP5. Neben den Befunden im Gastrointestinaltrakt können sich aber auch extrakolische Manifestationen entwickeln1.
Insgesamt kann die FAP innerhalb einer Familie sehr variabel verlaufen; dies betrifft nicht nur die Anzahl und das Manifestationsalter der Adenome, sondern auch die extraintestinalen Manifestationen11.
Epidemiologie
Ca. 5% aller kolorektalen Karzinome sind mit einem dominant oder rezessiv vererbten hereditären Syndrom assoziiert1. Am häufigsten ist hierbei das HNPCC auch Lynch Syndrom genannt1, gefolgt von der FAP, die ca. 1% aller kolorektalen Karzinom ausmacht1711. Sie tritt mit einer Prävalenz von ca. 1:10.000 auf. [1]257911.
Molekularpathogenese
Ursächlich ist eine Keimbahnmutation im APC Gen auf Chromosom 5q21-2278910. Dies ist bei 70%-80% der FAP-Patienten der Fall14511. De novo APC -Mutationen finden sich in bis zu 25% der FAP Fälle12789. Ca. 15-20% der Patienten mit einer scheinbaren de novo Mutation zeigen ein somatisches Mosaik12. Die klinische Bedeutung eines Mosaiks besteht darin, dass prädisponierte Kinder stärker als die Eltern betroffen sein können4. Daher sollte bei scheinbar neuen (sporadischen) Fällen mit klinisch unauffälligen Eltern immer nach einem Mosaik gesucht werden, da sich auch bei den Eltern unerkannt Adenome entwickeln können4.
Mutationen im APC Gen führen zu einer generalisierten Störung der Regulation des Gewebewachstums9. Das APC Gen ist ein Tumorsuppressorgen, das den Wnt-Signalweg hemmt79. Die Schlüsselkomponente in diesem Pathway nimmt ß-Catenin ein, welches die Transkription wachstumsregulatorischer Gene aktiviert7. Das APC -Genprodukt sorgt so für Downregulation von ß-Catenin79. Liegt eine Mutation vor, gelangen übermäßige Mengen an ß-Catenin in den Zellkern und stimulation so den Zellwachstum79. Das Resultat ist eine erhöhte Initiationsrate in der Adenom-Karzinom-Sequenz mit Ausbildung der Polyposis5910. Mutationen umfassen u.a. Insertionen, Deletionen, und nonsense Mutationen die zum Frameshift und/oder verfrühten Stopcodons führen8. Mehr als 500 Keimbahnmutationen, die ursächlich für die FAP sind, konnten gefunden werden, fast alle resultieren in einer Verkürzung des APC -Genproduktes9.
Da die FAP autosomal dominant vererbt wird, haben die Kinder betroffener Eltern ein 50%-tiges Risiko zu erkranken41011.
Das Auftreten und die Anzahl der Polypen ist abhängig von der Lokalisation der Mutation im APC -Gen18. Mutationen am Codon 1309 sind mit einer hohen Anzahl an Kolonpolypen assoziiert9, während Mutationen zwischen dem Codon 1250 und 1464 (zentral gelegen, „mutation-cluster-region“6 ) mit einer schweren Form der FAP einher gehen18, die früher manifest wird, und eher mit der Entwicklung eines kolorektalen Karzinoms vergesellschaftet ist9. Mutationen, die sich am Genende von Exon 9 befinden, sind hingegen mit einer milden Form der FAP assoziiert1. Eine mittelschwere Expression der Erkrankung findet sich bei Patienten mit Mutationen in den verbleibenden APC -Genarealen1.
Varianten der FAP
- MAP
Im Jahr 2002 beschrieb Al-Tassan et al. die Rolle der fehlerhaften BER in erblichen kolorektalen Tumoren1. Er identifizierte in einer britischen Familie mit drei betroffenen Mitgliedern und rezessiver Vererbung9 von mehreren kolorektalen Adenomen und Karzinomen eine biallele Keimbahnmutation im BER -Gen MUTYH110. Weitere Studien konnten biallele MUTYH -Mutationen in 26-29% der Patienten mit 10-100 Polypen und 7-29% der Patienten mit 100 – 1000 Polypen ausfindig machen1. Bei Patienten mit weniger als 10 Adenomen, und bei einigen mit einem kolorektalen Karzinom findet sich diese Mutation nur selten1.
Die biallelen MUTYH Mutationen sind in der Regel mit einem abgeschwächten Polyposis Phänotyp assoziiert1. Andere intestinale Tumoren und FAP -assoziierte extraintestinale Läsionen wie Duodenaltumoren, Osteome und CHRPE fanden sich bisher nur vereinzelt in MAP -Patienten1. Therapeutisch muss hier nicht zwangsläufig eine Kolektomie erfolgen, da die Patienten meist nur wenige Polypen haben1. Eine endoskopische Polypektomie ist hier u.U. ausreichend1. Patienten mit einer mono-allelen Mutation in MUTYH haben wahrscheinlich kein erhöhtes Risiko für ein kolorektales Karzinom und müssen daher nicht koloskopisch überwacht werden1.
Aufgrund der autosomal-rezessiven Vererbung findet sich eine positive Familienanamnese nicht über die Generation, sondern in der Generation (bei den Geschwistern)511.
Die klinische Differenzialdiagnose zu AFAP und HNPCC ist oft schwierig5. Die Erkrankung wird meist nach dem 45. Lebensjahr diagnostiziert5. In der Regel finden sich 10–100 Adenome, einige Patienten weisen jedoch mehr als 100 Adenome auf5. Bei bis zu 20% der Patienten findet sich auch eine Duodenalpolyposis5.
Die Indikation zum MYH-Gen-Test besteht beim Nachweis von mehr als 10 Adenomen15. Die MYH-Testung erfolgt derzeit meist bei FAP oder AFAP ohne Nachweis einer APC -Keimbahnmutation5. Insbesondere bei einem Stammbaum, der nicht zu einer autosomal-dominanten Erkrankung passt, ist an eine MYH-Testung zu denken5. Bei 10–20% der Patienten mit fehlendem Nachweis einer APC -Mutation und bei rund 2% aller Kolorektalkarzinom-Patienten mit Erstdiagnose vor dem 50. Lebensjahr können biallelsche MYH-Mutationen nachgewiesen werden5. Bisher sind keine spezifischen Vorsorgeempfehlungen für die MAP definiert5. Sie wird bislang nur für Träger von biallelschen Mutationen empfohlen5. Die Häufigkeit MUTYH -verursachter kolorektalen Karzinome liegt bei 1–2% aller kolorektaler Karzinome11 - AFAP
Ca. 8% der Familien mit FAP zeigen eine abgeschwächte Form, die sogenannte AFAP oder auch „attenuated form“ der FAP genannt, die mit weniger Adenomen einher und in der Regel später zu einem kolorektalen Karzinom führt1247. Das Lebenszeitrisiko ein kolorektales Karzinom zu entwickeln beträgt im Durchschnitt 69%2. Die Patienten haben im Schnitt ca. 30 Polypen (Variiert zwischen <5 zu 100 Polypen7), entwickeln tendenziell proximale Kolontumoren3579, bei seltenem Befall des Rektums7 und sind in der Regel älter als die klassischen FAP Patienten zum Zeitpunkt der Tumorentstehung279 (im Schnitt 55 Jahre3). Funduspolypen und duodenale Adenome finden sich häufig bei AFAP, CHRPE hingegen selten9.
Die Grundlage der AFAP ist wie bei der FAP eine Keimbahn-Mutation im APC -Gen2. Jedoch finden sich im Rahmen der AFAP nur in ca. 25% der Patienten Mutationen im APC -Gen1. Diese sind meist im 3´und 5´ Bereich des APC -Gens lokalisiert89.
Makroskopie der Polypen im Rahmen der FAP
Im Kolon und Rektum findet man unzählige (zwischen 100-1000) und unterschiedlich große Adenome13. Ihre Anzahl in den einzelnen Kolonabschnitten kann jedoch erheblich variieren8. Generell findet sich im linken Hemikolon eine höhere Dichte an Polypen als im rechten Hemikolon813. Auch die Größe der Polypen ist variabel13. Neben kaum sichtbaren mukösen Knötchen von 1-2mm bis hin zu 1cm großen oder oder noch größeren Polypen8 kann alles gefunden werden.
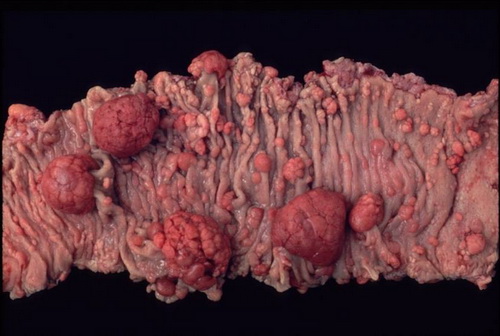
Abb. 521: Multiple Adenome bei FAP
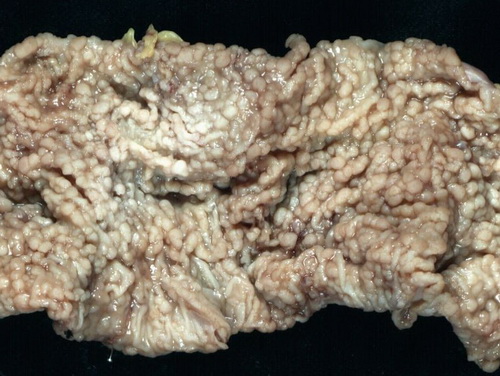
Abb. 511: Makroskopischer Aspekt des Kolon nach Aufschneiden und Formalinfixation (in Höhe des Colon ascendens). Multiple Polypen.
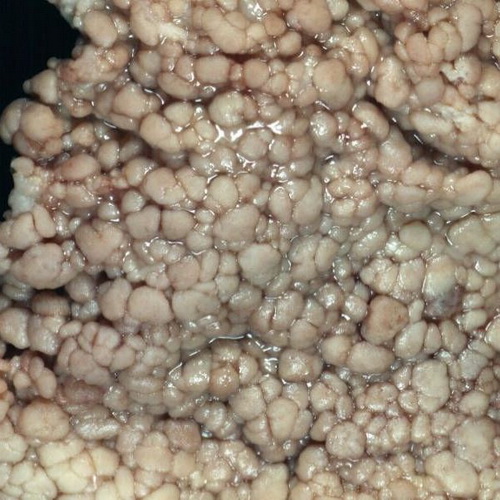
Abb. 560: Ausschnitt aus einem Kolektomiepräparat. Erkennbar sind zahlreiche Polypen unterschiedlicher Größe.
Die Adenomgröße ist alterskorreliert13. Zum Zeitpunkt der Krankheitsmanifestation mit etwa 15 Jahren findet man pfefferkorngroße breitbasig aufsitzende Protuberanzen13. Im weiteren Verlauf werden diese größer13. Weitehin finden sich tubuläre Adenome unterschiedlicher Größe auch im Dünndarm und im Magen13.
Es lassen sich drei Entwicklungsstadien der FAP gegeneinander abgrenzen13:
- ein sog. Latenzstadium bis zur Pubertät, i.allg. ohne nachweisbare Adenome
- das symptomatische Auftreten von Adenomen im jugendlichen Alter
- eine klinisch manifeste Phase, die etwa im 3. Lebensjahrzehnt beginnt.
Histologie
Histologisch dominieren bei FAP-Patienten überwiegend tubuläre Adenome13. Nur gelegentlich finden sich tubulovillöse Adenome; villöse Adenome sind selten8.
Einteilung der erblichen Tumorsyndrome
Traditionell werden erbliche Tumorsyndrome des Dickdarms eingeteilt in solche mit Polyposis (>100 Polypen) und ohne Polyposis (<3–5 Polypen)10.
In Abhängigkeit vom histologischen Bild erfolgt eine Unterteilung in adenomatöse (FAP, AFAP, MAP), hyperplastische/serratierte oder hamartomatöse Polypen (PJS, FJP, CS(Cowden-Syndrom))10.
Klinik
Die klinische Symptomatik der FAP ist uncharakteristisch13. Erste Beschwerden entwickeln sich für gewöhnlich nicht bevor eine Polyposis manifest ist8. Sie umfassen meist Veränderungen im Stuhlgang (Blut und / oder Schleim im Stuhl, Durchfall, Verstopfung8, Tenesmen13) in Kombination mit unspezifischen abdominalen Beschwerden4, Rückenschmerzen1 und Gewichtsverlust13. Das Ausmaß der Beschwerden steht offenbar in direkter Korrelation zur Karzinomentwicklung13. In verschiedenen Untersuchungsserien weisen wenigstens 50% der symptomatischen Patienten bereits ein Karzinom auf, während die Karzinomsequenz bei asymptomatischen Patienten weitaus geringer ist13.
Diagnostik
Endoskopie: Initial sollte eine flexible Sigmoidoskopie1 mit PE(Probeentnahme) und Histologie erfolgen8. Ist der Befund negativ, sollte die Untersuchung alle 2 Jahre wiederholt werden1. Finden sich hier jedoch Polypen, sollte eine komplette Koloskopie folgen1. Diese Untersuchung ist dann jährlich durchzuführen bis eine Proktokolektomie indiziert ist1.
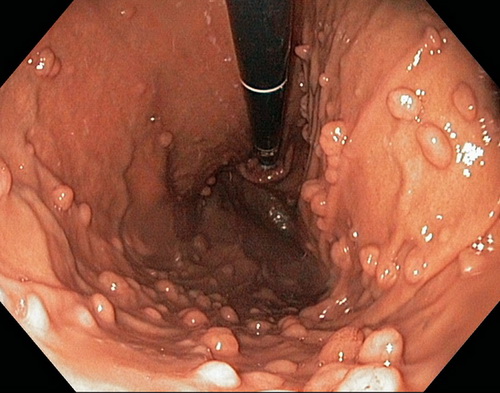
Abb. 613: Endoskopischer Aspekt mit multiplen kleinen Polypen (Quelle: http://www.endoskopiebilder.de/polypen_magen.html)
Bildgebung: Mit Hilfe der Computertomografie und der MRT ist es möglich, extrakolische Manifestationen wie z.B. abdominelle Desmoidtumoren nachzuweisen.
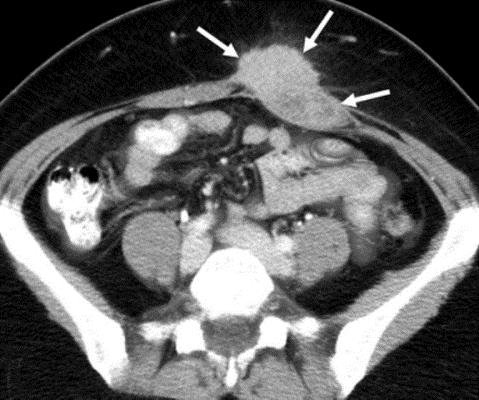
Abb. 614: Axiale CT-Aufnahme des Bauches mit Kontrastmittel. Erkennbar ist ein Desmoidtumor (Pfeile) mit Invasion des linken rectus abdominis. Obgleich Desmoidtumoren histologisch als benigne einzustufen sind, können sie ein lokal aggressives Wachstum zeigen. (Quelle: http://radiographics.rsna.org/content/25/6/1501.full)
Genetische Testung:
- Sequenzierung des gesamten APC -Gens8: Sehr sensitiver Test (95%-ige Mutationsdetektionsrate), aber auch sehr teuer7.
- Kombination aus CSGE und PTT : 80-90%-ige Mutationsdetektionsrate7.
- PTT : Wenn ein verkürztes Protein im Assay identifiziert wird, ist es möglich, die Mutation auf einem bestimmten Segment des Gens zu lokalisieren, um dann mit Hilfe der DNA -Sequenzierung die mutierten Nukleotide zu bestimmen8. 70-80%-ige Mutationsdetektionsrate7. Synonym IVSP8.
- Linkage analysis8: Sie findet Anwendung, wenn mehr als ein betroffenes Familienmitglied vorhanden ist (95-99%-ige Genauigkeit)7
Diagnose
Die Diagnose der FAP kann gestellt werden, wenn endoskopisch mehr als 100 kolorektale adenomatöse8 Polypen nachgewiesen werden128. Aber auch extrakolonische Manifestationen können auf eine FAP hinweisen2. Je mehr typische klinische und histologische Kriterien einer Polyposis nachweisbar sind, desto wahrscheinlicher ist es, dass eine Mutation vorliegt4.
Die Verdachtsdiagnose basiert somit auf klinischen und histologischen Faktoren und ist die Voraussetzung für eine gezielte Mutationsanalyse4. Diese erfolgt immer zuerst an einer bereits erkrankten Person, dem sogenannten Index-Patienten4. Ein negativer Mutationsbefund ist nicht gleichbedeutend mit dem Krankheitsausschluss, hier ist die klinische Diagnose ausschlaggebend49. Der Nachweis einer APC -Mutation bestätigt die Erkranung2. Ein positives Testergebnis macht es möglich, eine weitere prädiktive Diagnostik an klinisch gesunden Angehörigen vorzunehmen49. Die Testung sollte zwischen dem 12 und 15 Lj. erfolgen, damit rechtzeitig therapeutisch interveniert werden kann9. Eine frühere Testung ist in der Regel nicht erforderlich, da die Erkrankung zu diesem Zeitpunkt noch nicht klinisch manifest wird9.
Sind klinische Kriterien nicht erfüllt, sinkt die Mutationsdetektionsrate deutlich4. Mit der Einführung der MLPA, ist es mittlerweile möglich, große genomische Deletionen zu identifizieren, die das gesamte Gen oder einzelne Exons betreffen4. Durch dieses Verfahren erhöht sich die Mutationsdetektionsrate um 10% auf 30%4.
Liegt familiär eine gesicherte FAP vor, dann ist die Diagnose auch möglich, wenn weniger als 100 Polypen vorliegen5.
Die klinische Diagnose der AFAP ist hingegen schwieriger zu stellen1. Sie kann sowohl eine klassische FAP nachahmen als auch eine MAP und ein sporadisches Polypenwachstum imitieren2. Der Nachweis einer AFAP bei einer einzelnen Person kann aus diesem Grund schwierig sein2. Hier sollte eine Untersuchung der Familienmitglieder und ggf. die Identifikation des Phänotyps erfolgen2. Von einer AFAP kann ausgegangen werden, wenn mehr als 10 und weniger als 100 Polypen bei Personen zwischen dem 40. und 50. Lebensjahr nachweisbar sind2.
| Diagnostische Kriterien | |
|---|---|
| Nielsen et al. | Mindestens zwei Patienten mit 10-99 Adenomen und einem Alter >30 Jahre, ein Patient mit 10-99 Adenomen und einem Alter>30 Jahre und ein Verwandter ersten Grades mit einem kolorektalem Karzinom, basierend auf wenigen Adenomen1. Für beide Kriterien gilt, dass keine Familienmitglieder >100 Adenome vor dem 30. Lebensjahr haben dürfen1. |
| Knudson et al. | Dominante Vererbung der Erkrankung und 3-99 kolorektale Adenome mit 20 Jahren oder älter1. |
Andere genetisch bedingte gastrointestinale Erkrankungen mit Auftreten adenomatöser Polypen
Bei Vorkommen adenomatöser Polypen im Kolorektum oder allgemein im Gastrointestinaltrakt sind neben der FAP differentialdiagnostisch eine Reihe weitere, wesentlich seltenere Polyposis-Syndrome zu berücksichtigen. Diese lassen sich klinisch und/oder morphologisch gut voneinander abgrenzen. Hierbei spielen die Anzahl und die Verteilung der Polypen im Magen-Darm-Trakt, als auch die Histologie eine wichtige Rolle4. Aus diesem Grund basiert das initiale diagnostische Procedere bei einer Polypose immer auf einer Endoskopie mit Histologie in Zusammenschau mit extraintestinalen Manifestationen und der Familienanamnese.
| Attenuierte familiäre adonomatöse Polyposis (AFAP)4 | MUTYH (MutY Homolog von E.coli) assoziierte Polyposis4 | Hereditäres, nicht-Polyposis assoziiertes Kolorektalkarzinom (HNPCC)4 | Birt-Hogg Dubé syndrome (BHD)4 | |
|---|---|---|---|---|
| betroffenes Gen | APC5 | MUTYH | MLH1/2, MSH6, PMS2 | BHD (FLCN) |
| Häufigkeit | <1:10000 | <1:10000 | 1:500 | selten |
| Identifikation der Mutation in % | 20-305 | 15-20 (in APC negativen Patienten | 60-80 | 80-90 |
| Erbgang | Autosomal dominant | Autosomal rezessiv | Autosomal dominant | Autosomal dominant |
| Penetranz in % | ca. 100 | ca. 100 | ca. 80 | hoch |
| Anzahl der Polypen | 10-100 | 20-hunderte | 0->30 | vereinzelt bis multipel |
| Lokalisation der Polypen | Colon, Duodenum, Magen | Colon, Duodenum, Magen | Colon | Colon |
| Histologie | Adenom | Adenom | Adenom | Adenom |
| Extraintestinale Manifestationen | Selten | Erhöhte Inzidenz extraintestinaler Tumoren, selten Talgdrüsentumoren | Endometrium-, Tagdrüsen- und Magen-CA u.a. | Spezifische Haut- und Nierentumoren, Lungenzysten (Pneumothorax) |
| Lebenszeitrisiko in % ein kolorektales Karzinom zu entwickeln | 80-100 | 80-100 | ca. 80 | hoch |
Der Nachweis einer Mutation in Leukozyten-DNA ist für die Differentialdiagnose der Polyposis-Syndrome wichtig, dient aber auch der Bewertung des Risikos der Krankheitsentstehung (autosomal dominante vs. autosomal rezessive Vererbung), und der prädiktiven Testung gefährdeter asymptomatischer Personen4. Mit Hilfe prädiktiver Gentests können vorbeugende Maßnahmen bei Familienmitgliedern erfolgen die Mutationsträger sind4.
Weitere Manifestationen der FAP
- Intestinale Manifestationen:
Die Polypen können sich auch im oberen GI-Trakt entwickeln, hier vor allem im Duodenum1 aber auch im Magen.- Magen: Initial sollte eine flexible Sigmoidoskopie1 mit PE(Probeentnahme) und Histologie erfolgen8. Ist der Befund negativ, sollte die Untersuchung alle 2 Jahre wiederholt werden1. Finden sich hier jedoch Polypen, sollte eine komplette Koloskopie folgen1. Diese Untersuchung ist dann jährlich durchzuführen bis eine Proktokolektomie indiziert ist1.
- Duodenum: In 50-90% der Fälle finden sich Adenome im Duodenum145811. Das Lebenszeitrisiko duodenaler Adenome liegt bei nahezu 100%8. Sie kommen häufig an der Papilla Vateri589, periampullär38 und im mittleren und letzten Drittel des Duodenums zur Darstellung2. Die Anzahl und Größe variiert von wenigen, kaum sichtbaren bis hin zu über 100 Polypen8. Sie können als multiple diskrete Adenome (Durchmesser von 1-10mm) oder als flache konfluente Plaques in Erscheinung treten8. In manchen Fällen findet sich nur eine prominente Papille, oder die Mukosa erscheint blass mit weißlichen Belägen, die sich nicht abkratzen lassen8. Biopsien scheinbar normaler Mukosa zeigen regelmäßig Mikroadenome89.
Duodenale Tumoren finden sich am zweit häufigsten bei FAP bzw. AFAP -Patienten2. Das Lebenszeitrisiko beträgt zwischen 4%-12%2511. Es ist vor allem für Patienten mit schwergradiger Duodenalpolyposis und hohem Alter1 erhöht5. Eine Assoziation zwischen der Lokalisation der Mutation und der Anzahl der Polypen besteht nicht1. Dafür sind aber Mutationen downstream von Codon 1051 mit periampullären Adenomen assoziiert8. Sie entstehen in der Regel 15 Jahre später als die Kolonpolypen8. Duodenale Polypen werden nach der Spigelmann Classification eingeteilt18: - Spigelman Classification189
| Kriterium | 1 Punkt | 2 Punkte | 3 Punkte |
|---|---|---|---|
| Anzahl der Polypen | 1-4 | 5-20 | >20 |
| Polypengröße in mm | 1-4 | 5-10 | >10 |
| Histologie | tubulär | tubulo-villös | villös |
| Dysplasie | leicht | mittel | schwer |
| Stadium | Punktvergabe | Schwere der Erkrankung |
|---|---|---|
| 0 | Null Punkte | sehr mild |
| I | 1-4 Punkte | mild |
| II | 5-6 Punkte | mäßig |
| III | 7-8 Punkte | schwer |
| IV | 9-12 Punkte | sehr schwer |
- Ca. 80% der Patienten präsentieren sich im Stadium I – III, 10-20% im Stadium IV1. Prospektive Follow-up Studien konnten zeigen, dass sich die duodenalen Adenome wesentlich langsamer zum Krebs entwickeln als jene im Kolonrektum1. Da die Karzinogenese hier mit 15-20 Jahren angenommen wird, finden sich im Rahmen des Screenings die meisten duodenalen Adenome in einem Frühstadium1.
Das Risiko für die Entwicklung von Duodenaltumoren ist mit geschätzten 5% relativ gering, bei Patienten, die sich im Stadium IV nach Spiegelmann befinden, mit 7-36% jedoch weitaus höher19.
Möglichkeiten der lokalen Therapie duodenaler Adenome umfassen endoskopische Exzision58, Thermoablation8, Argon-Plasmakoagulation und die photodynamische Therapie1. Nach Brosens et al. ist das Rezidivrisiko (erneutes Polypenwachstum) nach endoskopischer Therapie erhöht, bei einer ebenfalls erhöhten Komplikationsrate1. (Perforation, Blutung, Pankreatitis). Eine operative Therapie ist indiziert bei villösen Polypen, Dysplasien, raschem Wachstum, und Verhärtungen8. Die Therapie der duodenalen Polypen beinhaltet zum einen lokale chirurgische Verfahren wie z.B.: die Duodenotomie mit Polypektomie und/oder Ampullektomie (beide Verfahren bergen erhöhte Komplikationen und eine erhöhte Rezidivrate8 ), aber auch ausgedehntere Verfahren wie die pankreaserhaltende Duodenotomie und die pyloruserhaltende Pankreatikoduodenektomie (Whipple-OP)159. Bei beiden Letztgenannten ist die Rezidivrate am geringsten1.
Bei Patienten mit Spigelman Stadium I ist aufgrund der geringen malignen Entartung und den erhöhten interventionellen Komplikationen lediglich ein Follow-up indiziert1. Patienten im Stadium III und IV, die jünger als 40 Jahre sind, sollten sich einer lokalen operativen Therapie, ältere Patienten einer Duodenketomie unterziehen1. Die operative Therapie ist dann sinnvoll, wenn im Duodenum nur 1-2 große Polypen bei sonst unauffälligem Befund nachweisbar sind1. Ca. 20% der Patienten müssen sich im Verauf einer therapeutischen Endoskopie oder einer Operation unterziehen2.
Das endoskopische Screening auf duodenale Polypen sollte zwischen dem 25.-30. Lebensjahr erfolgen19. - Vorsorgeintervalle nach der Spigelman Klassifikation1:
| Spigelman Klassifikation | Vorsorgeintervall (Jahren) |
|---|---|
| Stadium I und ggf. II | 5 |
| Stadium II | 3 |
| Stadium III | 1-2 |
| Stadium IV | Chir. Therapie erwägen |
- Extraintestinale Manifestationen:
- Desmoidtumoren: Sie entwickeln sich bei 10-15%245 der FAP-Patienten. Bei AFAP-Patienten sind sie hingegen seltener2. Histologisch stellen sie benigne18 klonale Proliferationen von Myofibroblasten1011 dar. Ein Metastasierungspotential besteht nicht89. Nichts desto trotz können sie aufgrund ihres Wachstumsverhaltens (diffus-infiltrierend vs. solide9 ) mit Ummauerung großer Gefäße oder Kompression umgebender Strukturen289 einen malignen Charakter annehmen, und sollten nach Möglichkeit therapiert werden1.
Risikofaktoren für die Entstehung von Desmoiden sind das weibliche Geschlecht, insbesondere hohe Östrogenspiegel (Schwangerschaft oder hormonelle Kontrazeption), eine positive Familienanamnese für Desmoide, das Vorliegen von Osteomen, intraabdominelle chirurgische Eingriffe11 und die Lokalisation der APC-Mutation1. Abdominelle Desmoidtumoren finden sich bevorzugt bei Patienten mit Mutationen zwischen den Codons 14449/1445 und 15788. Sie können aber auch bereits kongenital vorliegen11.
Im Vergleich zu sporadischen Tumoren finden sich die FAP assoziierten Tumoren überwiegend im Bereich der Bauchwand (15%)9, intraabdominal, hier besonders im kleinen Netz (70%)19, in Operationsnarben oder extraabdominal (Brustwand oder intrathorakal)8. Ihr Wachstumsverhalten variiert von raschem Progress einhergehend mit Symptomen aufgrund Kompression viszeraler Organe, bis hin zu einem langsamen Verlauf; auch spontane Regressionen konnten nachgewiesen werden8.
Leitsymptome sind schmerzhafte (50%) bzw. schmerzlose abdominelle Raumforderungen8. Die Schmerzen entstehen für gewöhnlich durch die Kompression von Darmanteilen (Ischämie) mit dem Risiko einer Perforation9, durch eine Ureterobstruktion, oder Einblutungen in den Tumor8. Die Diagnostik der Wahl beinhaltet das CT und das MRI8. Die Bildgebung erlaubt die exakte Größenbestimmung der Tumoren und ihre Lage zu Nachbarorganen9.
Charakteristische histologische Befunde beim Desmoidtumor sind in der Rosai Collection zu finden.
Nichtchirurgische therapeutische Optionen umfassen zum einen die Gabe von NSAID wie z.B.: Sulindac und/oder die Gabe von Antiöstrogenen (Tamoxifen)5, zum anderen kann eine Chemo- oder Radiotherapie in Erwägung gezogen werden1. Weiterhin kann eine operative Entfernung erfolgen. Dabei gilt es jedoch zu beachten, dass jeder chirurgische Eingriff ein Wachstumsreiz für weitere Desmoide darstellt, bzw. es bei unvollständiger Resektion zum erneuten Wachstum der Tumoren kommt189. Eine Operation sollte daher nach Möglichkeit nur bei lebensgefährlichen Komplikationen in Betracht gezogen werden8. Ziel sollte es sein, den chirurgischen Eingriff so weit wie möglich hinauszuzögern. Viele Patienten müssen jedoch im weiteren Krankheitverlauf operiert werden8. Sind die Tumoren noch sehr klein, können bei großzügiger Resektion viele Patienten geheilt werden8.
Nach prophylaktischer Kolektomie stellen Desmoide und Karzinome des oberen Gastrointestinaltrakts heutzutage die häufigsten krankheitsassoziierten Todesursachen der FAP dar5. Sie können zum einen durch Erosion in Blutgefäße zum anderen durch eine Sepsis aufgrund von Fistelbildung, aber auch in Folge von Operationskomplikationen zum Tode führen8. Zwischen 2 und 5% aller FAP -Patienten versterben an den Folgen von Desmoiden. - Staging für das Management von Desmoidtumoren nach Church et al.8
- Desmoidtumoren: Sie entwickeln sich bei 10-15%245 der FAP-Patienten. Bei AFAP-Patienten sind sie hingegen seltener2. Histologisch stellen sie benigne18 klonale Proliferationen von Myofibroblasten1011 dar. Ein Metastasierungspotential besteht nicht89. Nichts desto trotz können sie aufgrund ihres Wachstumsverhaltens (diffus-infiltrierend vs. solide9 ) mit Ummauerung großer Gefäße oder Kompression umgebender Strukturen289 einen malignen Charakter annehmen, und sollten nach Möglichkeit therapiert werden1.
| Stadium | Wachstumsverhalten | Management |
|---|---|---|
| I | Asymptomatisch, nicht wachsend | Sie sind meist ein Zufallsbefund im Rahmen der Bildgebung oder während der OP, und sollten bei kleinen Befunden beobachtet werden. Neben der Verlaufsbeobachtung werden häufig NSAID gegeben. Sind die Tumoren noch klein, und können ohne ausgedehnte Resektion entfernt werden, sollte eine Operation erfolgen. |
| II | Symptomatisch, max. Durchmesser 10cm oder kleiner, nicht wachsend | Kleine Desmoide, die symptomatisch werden, bedürfen in jedem Fall einer Operation, sofern diese möglich ist, auch wenn sie nicht am wachsen sind. Ist eine Operation nicht mehr möglich, können NSAID in Kombination mit Tamoxifen und/oder Raloxifen gegeben werden. |
| III | Symptomatisch, Durchmesser zwischen 11cm und 20cm, oder asymptomatisch und langsam wachsend (<50% Zunahme des Durchmessers in 6 Monaten) | Diese Tumoren bedürfen einer aktiven Behandlung. NSAID, Tamoxifen, Raloxifen und Vinblastin/Methotrexat sind hier die Mittel der Wahl. Wächst der Tumor weiter, kann eine antisarkomatöse Chemotherapie gegeben werden. |
| IV | Symptomatisch, Durchmesser >20cm, schnell wachsend (>50% Zunahme des Durchmessers in 6 Monaten), Auftreten von Komplikationen | Dieses Stadium birgt die meisten Komplikationen wie Sepsis, Perforation oder Blutungen. Hier sollte dringend therapiert werden. Neben Exenteration, antisarkomatöser Therapie spielt hier auch die Bestrahlung eine wichtige Rolle. |
- Osteome: Sie sind häufig an der Schädelkalotte und der Mandibula lokalisiert245711, aber prinzipiell auch an jedem anderen Knochen zu finden8. Trotz Benignität wachsen sie häufig lokal und werden so symptomatisch8. Osteome werden in manchen Fällen vor der Diagnose FAP entdeckt811.
- CHRPE2478: Hierbei handelt es sich um gutartige Pigmentveränderungen der Netzhaut (histologisch Harmatome8 ), deren Auftreten mit einer Mutation im Codon 450/463-1444 assoziiert ist89. Mit Hilfe der indirekten Ophthalmoskopie kann die runde bis oval pigmentierte Läsion mit einem zirkulären hellen Halo diagnostiziert werden8. Die Inzidenz bei Patienten mit FAP variiert zwischen 50-79%8. Die Sensitivität der CHRPE in FAP -Patienten beträgt 79%, die Spezifität 95%8. Das Fehlen einer CHRPE ist nicht gleichbedeutend mit dem Fehlen der FAP8. Die Läsionen können bereits bei Geburt vorhanden sein, und führen in der Regel nicht zu einer Visuseinschränkung [11]. Einzelne Harmatome werden auch in der Normalbevölkerung beobachtet11.
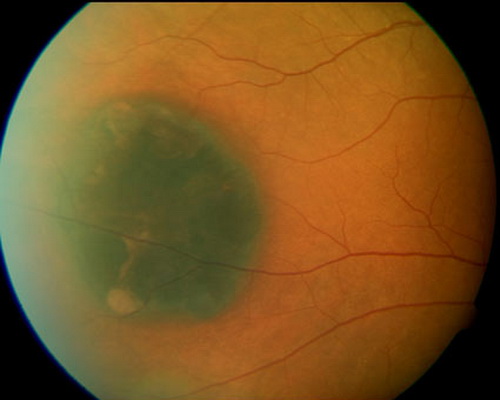
Abb. 615:
Typisches Bild einer solitären kongenitalen Hypertrophie des retinalen Pigmentepithels. Die Läsion ist tiefschwarz pigmentiert, ist scharf begrenzt und es fallen depigmentierte Lakunen innerhalb der Veränderung auf. (Quelle: http://www.medizin.uni-tuebingen.de/Presse_Aktuell/Einrichtungen+A+bis+Z/Kliniken/Augenheilkunde/Augenklinik/Sprechstunde+f%C3%BCr+intraokulare+Tumore/Kongenitale+Hypertrophie+des+retinalen+Pigmentepithels+(CHRPE).html)
- Epidermoidzysten2457 und Fibrome27 : Sie zählen zu den gutartigen kutanen Veränderungen und finden sich in den Extremitäten, im Gesicht und an der Kopfhaut8. Die Zysten können schon vor Entwicklung der kolorektalen Polypen in Erscheinung treten8.
- Zahnanomalien: Es finden sich u.a. noch nicht durchgebrochene Zähne, ein angeborenes Fehlen der Zahnanlage2, oder überzählige Zähne7. Daneben können auch verschmolzene Zahnwurzeln der ersten und zweiten Molaren und ungewöhnlich lange Wurzelanlagen in Erscheinung treten8. Die Zähne können in untypischer Lokalisation (z. B. Gaumen) durchbrechen, sie können aber auch als zystische Degeneration im Kieferknochen liegen11. Es sind zwischen 11 und 80% aller Patienten mit FAP betroffenen8. Auch diese Anomalien können schon vor der FAP diagnostisch in Erscheinung treten8.
Gardner-Syndrom: Phänotypische Variante der FAP5. Als Gardner-Syndrom wird eine FAP mit Mutationsnachweis im APC-Gen, dem vermehrten Auftreten von Weichteiltumoren (Epidermoidzysten, Fibrome und Desmoide), Osteomen, Zahnanomalien und einer Polypose des oberen Gastrointestinaltrakts bezeichnet. [8]911. Bei Patienten mit sehr ausgeprägtem Auftreten von Weichteiltumoren wird die Bezeichnung “Gardner-Syndrom” noch verwendet11, ansonsten ist der Begriff historisch8. Der histologische Befund beim Gardner-Syndrom (hier: 11 Jahre alter männl. Patient mit rektalen Blutungen und epidermalen Einschlußzysten) ist gleichartig wie bei der FAP.
Turcot-Syndrom: Das Turcot Syndrom wurde ursprünglich in zwei Geschwistern mit Polyposis coli beschrieben, die bösartigen Hirntumoren entwickelt haben12. Diese seltene Erkrankung wird wahrscheinlich autosomal-dominant vererbt12. Die Häufigkeit des Turcot Syndroms ist schwer zu beurteilen, da die Tumoren des Gehirns mit einer hohen Mortalität assoziiert sind, und dem Nachweis von Kolonpolypen vorausgehen können12. Studien konnten folgende zwei Formen des Turcot Syndroms: eine Gruppe bestehend aus Patienten mit Gliomen und kolorektalen Adenomen (Non-Polyposis-Patienten) und eine weitere Gruppe bestehend aus Patienten mit Hirntumoren (vorwiegend Medulloblastomen) mit adenomatöser Polyposis891112. Molekulare Untersuchungen konnten zeigen, dass in der erste Gruppe vor allem Keimbahn-Mutationen von Mismatch-Reparatur-Genen (hMLH1 und hPMS2) vorliegen und somit Teil der erblichen Non-Polyposis-Syndrome darstellt, während in der letzteren Gruppe eine Keimbahn-Mutation des APC-Gens zugrunde liegt12.
Muir-Torre-Syndrom: Das Muir-Torre-Syndrom ist eine autosomal-dominant vererbte Erkrankung mit einem variablen Phänotyp12. Es finden sich vorwiegend Hauttumoren mit talgdrüsenartiger Differenzierung und viszerale Malignome12. Fünfzig Prozent der Patienten haben Darmkrebs und 15% der weiblichen Patienten entwickeln ein Endometriumkarzinom12. Muir-Torre-Patienten zeigen Mutationen ähnlich denen des hereditären Polyposis-Syndrom (HNPCC)12.
- Tumormanifestationen:
- Kolon7: Lebenszeitrisiko von 100%2
- Duodenum7: Lebenzeitrisiko von 4-12%2
- Magen7: Lebenszeitrisiko <1%2
- Pankreas7: Lebenszeitrisiko 2%2311
- Schilddrüse7: Papilläres Schilddrüsenkarzinom234; Lebenszeitrisiko 1-2%211, bevorzugt Frauen betroffenen11.
- Leber7: Lebenszeitrisiko 1-2% ein Hepatoblastom zu entwickeln23411 Ein Hepatoblastom kann im Kindesalter die Primärmanifestation einer FAP darstellen11.
- ZNS7: Lebenszeitrisiko <1% ein Medulloblastom im Kleinhirnbrückenwinkel zu entwickeln23411.
- Gallenblase und Gallenwege79: Lebenszeitrisiko <1%11.
Therapie
Operative Therapie: Die prophylaktische Proktokolektomie im jungen Erwachsenenalter (16.-20. Lebensjahr9) und in einem prämalignen Stadium führt maßgeblich zur signifikaten Reduktion der Mortalität und Morbidität, welche mit einem fortgeschrittenen kolorektalen Karzinom einhergeht1. Sie sollte in Erwägung gezogen werden, wenn mehr als 20 Adenome nachweisbar sind, diese >1cm messen oder bei suspekter Histologie2.
Man unterscheidet vor allem zwei operative Methoden1, die beide laparoskopisch durchgeführt werden können9:
| IRA | IPAA. | |
|---|---|---|
| OP-Verfahren/Indikation | Die IRA ist eine relativ einfache Operation mit niedriger Komplikationsrate (keine Plexusverletzung9 ) , gutem postoperativem Ergebnis178 und einem kürzeren Klinikaufenthalt9. Mit dieser Methode werden bevorzugt junge Patienten mit wenig Adenomen (<20 rektale Adenome, <1000 Adenome im Kolon) und/oder Mutationen im Codon 0-200 oder >1500 operiert8. Ein Nachteil der Methode ist die Gefahr der Karzinomentwicklung im verbleibendem Rektum, welche abhängig von der Anzahl der dortigen Polypen ist9. Das Risiko ein Rektumkarzinom zu entwickeln steigt, wenn mehr als 20 Polypen im Rektum vorliegen8 und beträgt dann zwischen 12%-29% in den nächsten 20-25 Jahren9. Vermehren sich die Polypen im Rektum und kann eine sichere Entfernung nicht mehr gewährleistet werden, sollte eine Proktektomie erfolgen8. | Die IPAA ist im Vergleich ein weitaus ausgedehnteres operatives Verfahren, das vermehrt mit Blutungen, Infertilität, Nervenschäden im kleinen Becken1 aber auch mit Sepsis und Inkontinenz8 einher gehen kann. Sie sollte daher nicht bei Frauen mit Kinderwunsch angewendet werden1. Sie ist das Verfahren der Wahl bei einer hohen Anzahl an rektalen Adenomen (>15-20)12 und eliminiert nahezu das Risiko ein kolorektales Karzinom zu entwickeln7. Bei dieser Methode verbleiben noch ca. 1-2cm der rektalen Mucosa2. Finden sich hier auch Adenome, kann ein Mucosal stripping erfolgen2. Daneben können sich aber auch Adenome und Karzinome im Ilioanalen Pouch und im Bereich der Anastomose entwickeln8. Die primäre IPAA ist indiziert bei Patienten mit einem erhöhtem Risiko für ein Desmoidtumor1. |
| Follow-up | Die Frequenz des Follow-up des Rektums nach einer IRA ist abhängig von der verbleibenden Zahl an rektalen Polypen1. Die Intervalle variieren zwischen 6 und 12 Monaten89. Bei Patienten mit großen (>5mm) rektalen Adenomen mit hohem Dysplasiegrad wird eine Proktektomie empfohlen1. Methode der Wahl ist die flexible Endoskopie. | Auch bei Patienten mit einer IPAA sollte ein Follow-up erfolgen, da sich Adenome und Tumore in der verbliebenen Rektummukosa2, im Bereich der Anastomose2 oder im Pouch1 entwickeln können. Die Intervalle betragen hier zwischen 6-12 Monate1. Methode der Wahl ist auch hier die flexible Endoskopie9. |
Die rechtzeitige Proktokolektomie ist zur Karzinomprävention bei FAP entscheidend5. Es gibt keine Guidelines, die besagen, ab wann eine chirurgische Intervention zu erfolgen hat1. Generell wird eine (Prokto -) Kolektomie ab dem Auftreten einer großen Anzahl an Adenomen, und einer Größe >5mm und einem erhöhten Dysplasiegrad empfohlen1. Die meisten Patienten mit einer FAP werden zwischen dem 15. und 25. Lebensjahr operiert15.
Die Wahl der Operationsmethode hängt unter anderem vom Alter der Patientin, der Anzahl der rektalen Polypen, dem Kinderwunsch, dem Risiko für die Entwicklung von Desmoiden und der Lokalisation der Mutation im APC -Gen ab1.
Ca. 33% der Patienten, die rektumerhaltend operiert wurden, müssen sich im weiteren Verlauf aufgrund diffuser Polypen einer Proktektomie unterziehen2.
Medikamentöse Therapie: Das erste Medikament, das positive Effekte auf Patienten mit FAP hat, ist Sulindac12 ein NSAID9. Eine Dauertherapie reduziert die Zahl der Adenome um mehr als 50% im Kolon als auch im verbliebenen Rektumstumpf nach Kolektomie1. Die Entwicklung von Adenomen kann jedoch nicht verhindert werden1. In der postoperativen Prophylaxe kann es zusammen mit der Endoskopie im Rahmen der Nachsorge angewendet werden129. Da Sulindac in Deutschland nicht zugelassen ist, kann alternativ Indometacin substituiert werden14. Die Ergebnisse hierbei sind jedoch sehr viel schlechter als bei der Therapie mit Sulindac14.
Eine weitere medikamentöse Option ist die Gabe von Celecoxib, ein selektiver Cyclooxygenase-2-Hemmer9. Hier konnte in einer Studie bei zweimaliger Gabe pro Tag über einen Zeitraum von 6 Monaten eine Reduktion an Polypen um 28% gezeigt werden3. Auch duodenale Polypen reduzieren sich unter der Gabe von Celecoxib8. Allerdings können die Polypen unter den antiinflammatorischen Medikamenten wieder wachsen3. Diese Therapie ersetzt aus diesem Grund auf keinen Fall das endoskopische Screening, auch wenn es die prophylaktische Kolektomie verzögern mag3.
Beide Medikamente können die Karzinomentstehung nicht verhindern9.
Vorsorge
- FAP:
Bei FAP-Patienten und ihren Angehörigen ist eine regelmäßige Vorsorge durchzuführen5. Das Alter, bei dem mit der Vorsorge begonnen werden sollte, ist abhängig vom Risiko der malignen Transformation der kolorektalen Adenome1. In der aktuellen Literatur gibt es hierzu keine direkten Angaben1. Eine Empfehlung basiert auf dem Beginn des Screenings im Alter von 10-12 Jahren23. Hier ist sowohl bei unklarem Mutationsstatus als auch bei nachgewiesener familiärer Mutation jährlich eine Rektosigmoidoskopie indiziert579. Daneben kann gerade bei unbekanntem Mutationsstatus eine genetische Testung erforderlich werden7. Das Risiko ein kolorektales Karzinom zu entwickeln ist vor dem 20. Lebensjahr sehr gering1. Hier sollte im Abstand von 1-2 Jahren eine Koloskopie erfolgen2. Bei erstmaligem Nachweis von Adenomen ist nachfolgend jährlich eine Koloskopie durchzuführen257. Bei Entwicklung einer floriden Polyposis sollte eine Proktokolektomie erfolgen1. Dies ist meist im späten Teenageralter der Fall3. Patienten mit einer Mutation auf dem Codon 1309 im APC Gen können schon multiple Polypen vor dem 10. Lebensjahr entwickeln, und sollten bei Auftreten von Symptomen frühzeitig koloskopiert werden1. Bei Verwandten ersten Grades, bei denen keine Mutation gefunden wurde, sollte die Vorsorge mind. bis zum 50. Lebensjahr fortgeführt werden1. Die Vorsorgemaßnahmen führen zu einer Verringerung der Todesrate an kolorektalem Karzinom bei FAP-Patienten1. Ist bei einem Patienten eine FAP genetisch gesichert, sollte allen Verwandten ersten Grades eine genetische Beratung empfohlen werden7.
Im Einzelfall, insbesondere bei früher Adenom-/Karzinommanifestation (vor dem 10. Lebensjahr), kann die molekulargenetische (Blut-)Testung zur Bestimmung des Schweregrades der FAP und damit auch zur Planung des Operationszeitpunktes und der Überwachungsstrategie herangezogen werden10.
Insbesondere bei unklaren Fällen (<100 Polypen, höheres Manifestationsalter der Tumoren) empfiehlt es sich, die beim betroffenen Patienten aufgetretenen Tumoren genau auf mögliche Zugehörigkeit zum Tumorspektrum einer FAP(Familiäre adenomatöse Polyposis) zu prüfen10. So können scheinbar harmlose Fundusdrüsenpolypen, wenn diese z. B. als „multiple Drüsenkörperzysten“ im proximalen Magen eines jüngeren Patienten auftreten, den ersten Hinweis auf eine mögliche FAP geben10. In solchen Fällen sollte eine weitergehende koloskopische Abklärung empfohlen werden10.
Eine Erstuntersuchung des oberen Gastrointestinaltraktes ist spätestens bis zum 30. Lebensjahr durchzuführen (Eine Indikation besteht ab dem 25. Lebensjahr1) (Empfehlung des NCCN )58. Bei unauffälligem Befund der ÖGD(Ösophagogastroduodenoskopie) wird ein Intervall von 3 Jahren empfohlen25. Bei Nachweis von Adenomen ist das Intervall in Abhängigkeit vom Schweregrad der Duodenalpolypen auf 1–2 Jahre zu verkürzen5. Gerade Duodenaladenome sollten im Eingangsgut zur Überprüfung von Alter (<50 Jahre?) und ggf. weiterer Tumoren Anlass geben10. Diese treten häufiger später im Verlauf einer FAP, nicht selten erst nach der Tumorkolektomie auf10.
Ab dem 10.– 12. Lebensjahr ist jährlich eine Abdomen- und Schilddrüsensonographie durchzuführen25. Dies gilt sowohl für FAP als auch für AFAP-Patienten2 - AFAP:
Bei Patienten mit AFAP entwickeln sich die Adenome vorzugsweise im rechten Kolon; aus diesem Grunde sollte im Rahmen der Vorsorge immer eine Koloskopie erfolgen1. Als Vorsorge wird eine erste Koloskopie im 15. Lebensjahr empfohlen, dann ab dem 20. Lebensjahr jährlich, da kasuistisch auch von kolorektalen Karzinomen bei jungen Patienten berichtet wurde5. Ca. 33% der Patienten mit einer AFAP sind über einen langen Zeitraum mit regelmäßigen Koloskopien und Polypektomien ausreichend versorgt2. Der Rest muss sich im Laufe der Zeit einer Kolektomie unterziehen2. Diese ist nur bei endoskopisch nicht beherrschbarer Polyposis indiziert5.
Auch die Endoskopie incl. side viewing Endoskopie im oberen GI-Trakt sollte bei der AFAP wie bei der FAP alle 1-3 Jahre erfolgen2. Begleitend sollte ein endoskopischer Ultraschall aller auffälliger Areale im Bereich der Ampulle durchgeführt werden2. Auch diese Untersuchungen sollten bereits zwischen dem 25. und 30. Lebensjahr erfolgen2.
Extrakolonische Manifestationen (Schilddrüse und Abdomen) können wie bei der klassischen FAP auftreten, daher sind weitere Untersuchungen wie bei der FAP s.o. durchzuführen5.
Prognose
Bei symptomatischen FAP-Patienten ist die Inzidenz eines kolorektalen Karzinoms mit 50-70% wesentlich höher als bei den Patienten die im Rahmen der Vorsorge auffallen (Inzidenz von 3-10%)1. Erfolgt keine Prävention, liegt das Kolorektalkarzinom-Risiko mit 21 Jahren bei 7%, mit 45 Jahren bei 87% und mit 50 Jahren bei 93%11. Das Intervall zwischen dem Auftreten der ersten klinischen Symptome und der oft multizentrischen Karzinomentstehung wird in der Literatur mit 10-15 Jahren angegeben13. Das kolorektale Karzinom manifestiert sich auf dem Boden einer FAP in etwa 20 Jahre früher als das kolorektale Karzinom ohne vorbestehende Adenomatose13.
Patienten mit einer FAP haben ein erhöhtes Risiko für die Entwicklung von Tumoren in anderen Organen, z.B. In der Schilddrüse (das Risiko ist um den Faktor 20,9 erhöht13, im Gehirn und in der Leber1. Die Haupttodesursachen im Rahmen der FAP sind jedoch Desmoide und Tumoren des Duodenums (das Risko, ein periampulläres Karzinom zu entwickeln ist um den Faktor 250 erhöht13 )1.
Nicht-adenomatöse Polyposis-Syndrome des Kolorektum
Differentialdiagnostisch muss die FAP abgegrenzt werden von folgenden seltenen, familiär und nicht-familiär auftretenden, nicht-adenomatöse Polyposen [11]. Zu erwähnen sind hier die serratierte Polypose, die juvenile Polypose und die lymphomatöse Polypose des Kolorektum. Zur differentialdiagnostischen Abgrenzung gegenüber der FAP siehe folgende Tabelle mit den diagnostische Kriterien der klassischen und attenuierten FAP im Vergleich zu anderen, nicht-adenomatösen kolorektalen Polyposen11
| Polypose | Diagnostische Kriterien |
|---|---|
| Klassische familiäre adenomatöse Polyposis (FAP) | > 100 Adenome, frühe klinische Manifestation (typisch) |
| Attenuierte FAP (AFAP) | > 10 bis 100 kolorektale Adenome oder > 100 Adenome bei später klinischer Manifestation (> 45. Lebensjahr) |
| Familiäre juvenile Polyposis | > 5 juvenile Polypen (JP) im Kolorektum oder ein und mehr JP bei positiver Familienanamnese für familiäre juvenile Polyposis |
| Serratierte Polyposis | mindestens 5 histologisch gesicherte hyperplastische Polypen proximal des Sigmas, mindestens zwei davon > 1 cm oder jede Zahl hyperplastischer Polypen proximal des Sigmas bei erstgradig Verwandten mit hyperplastischer Polyposis oder > 20 bis 30 hyperplastische Polypen über das Kolon verteilt |
| Lymphomatoide Polypose | Lymphomatöse Polypen beim primären Mantelzell- oder anderen NHL-Lymphomen des Kolorektum |
Dabei stellt die lymphomatöse Polypose eine seltene Form des gastrointestinalen NHL dar, repräsentiert in erster Linie durch das Mantelzell-Lymphom (MCL). Aber auch follikuläre Lymphome und MALT-Lymphome können vorkommen17. Makroskopisch können die kleine polypöse Tumoren auch an anderen Stellen des GI-Traktes vorkommen und treten in Clustern auf. Histologisch sind die Polypen mit Lymphomzellen in der Mukosa oder der Submukosa durchsetzt17.
Weiterführende Literatur
Guidelines for the clinical management of familial adenomatous polyposis (FAP)
H. F. A. Vasen et al.; Guidelines for the clinical management of familial adenomatous polyposis (FAP), Gut 2008;57;704-713Hereditary and Familial Colon Cancer
K.W. Jasperson, T.M. Tuohy, D.W. Neklason, and R. W. Burt; Hereditary and Familial Colon Cancer, Gastroenterology. 2010 June ; 138(6): 2044–2058.Hereditary Colorectal Cancer
H. T. Lynch, M.D. and A. de la Chapelle, M.D., Ph.D., Hereditary Colorectal Cancer, N Engl J Med 2003;348:919-32.The Differential Diagnosis and Surveillance of Hereditary Gastrointestinal Polyposis Syndromes
S. Aretz, The Differential Diagnosis and Surveillance of Hereditary Gastrointestinal Polyposis Syndromes, Dtsch Arztebl Int 2010; 107(10): 163–73Polyposis-Syndrome und Dünndarmkarzinome
Bernhardt C, Schulmann K, Schmiegel W. Polyposis-Syndrome und Dünndarmkarzinome. Der Gastroenterologe. 2009. 4:35–41.Richtlinien zur prädiktiven genetischen Diagnostik
Richtlinien zur prädiktiven genetischen Diagnostik (verabschiedet vom Vorstand der Bundesärztekammer am 14. 2. 2003); Deutsches Ärzteblatt, Jg. 100,Heft 19,9. Mai 2003Differenzialdiagnostik erblicher Dickdarmkarzinomsyndrome
Rüschoff J, Heinmöller E, Hartmann A, Büttner R, Rau T. Differenzialdiagnostik erblicher Dickdarmkarzinomsyndrome. Der Pathologe. 2010. 31:412–422.Familiäre adenomatöse Polyposis und andere Polyposissyndrome
Holinski-Feder E, Morak M. Familiäre adenomatöse Polyposis und andere Polyposissyndrome. Der Gastroenterologe. 2010. 5:7–15.Hereditäre nicht-polypöse kolorektale Karzinome
Holinski-Feder E, Morak M. Hereditäre nicht-polypöse kolorektale Karzinome. Deutsche Medizinische Wochenschrift. 2008 Aug;133(33):1690-5.Hyperplastische Polypen, sessile serratierte Adenome, konventionelle Adenome: Molekulare Pathways und deren klinische Relevanz
E. Holinski-Feder, M. Morak: Hyperplastische Polypen, sessile serratierte Adenome, konventionelle Adenome: Molekulare Pathways und deren klinische Relevanz. J Gastroenterol Hepatol Erkr 2010; 8 : 18 –25.
Referenzen
1 H. F. A. Vasen et al.; Guidelines for the clinical management of familial adenomatous polyposis (FAP), Gut 2008;57;704-713
2 K.W. Jasperson, T.M. Tuohy, D.W. Neklason, and R. W. Burt; Hereditary and Familial Colon Cancer, Gastroenterology. 2010 June ; 138(6): 2044–2058.
3 H. T. Lynch, M.D. and A. de la Chapelle, M.D., Ph.D., Hereditary Colorectal Cancer, N Engl J Med 2003;348:919-32.
4 S. Aretz, The Differential Diagnosis and Surveillance of Hereditary Gastrointestinal Polyposis Syndromes, Dtsch Arztebl Int 2010; 107(10): 163–73
5 C. Bernhardt, K. Schulmann, W. Schmiegel, Polyposis-Syndrome und Dünndarmkarzinome, Gastroenterologe 2009 · 4:35–41
6 Richtlinien zur prädiktiven genetischen Diagnostik (verabschiedet vom Vorstand der Bundesärztekammer am 14. 2. 2003); Deutsches Ärzteblatt, Jg. 100,Heft 19,9. Mai 2003
7 J. H. Scholefield, A. Grothey, H. Abcarian, T.Maughan, Challenges in Colorectal Cancer, Second edition 2006, Blackwell Publishing Ltd.
8 P. H. Gordon, S. Nivatvongs Principles and Practice of Surgery for the Colon, Rectum, and Anus, Third Edition, 2007, Informa Healthcare USA, Inc.
9 B. G. Wolff, MD, J. W. Fleshman, MD, D. E. Beck, MD, S. D. Wexner, MD, J. H. Pemberton, MD, The ASCRS Textbook of Colon and Rectal Surgery, 2007 Springer Science+Business Media, LLC
10 J. Rüschoff, E. Heinmöller, A. Hartmann, R. Büttner, T. Rau; Differenzialdiagnostik erblicher Dickdarmkarzinomsyndrome – Der Beitrag der Pathologie; Pathologe 2010; 31:412–422
11 E. Holinski-Feder, M. Morak; Familiäre adenomatöse Polyposis und andere Polyposissyndrome; Gastroenterologe 2010; 5:7–15
12 S. E. Mills, D. Carter, J. K. Greenson, und V. E. Reuter, Sternberg’s Diagnostic Surgical Pathology, Lippincott Williams & Wilkins, Auflage: 4th revised edition
13 W. Remmele, Pathologie Bd. 2 Verdauungstrakt, Springer-Verlag Berlin Heidelberg, 2. Auflage 1996
14 http://de.wikipedia.org/wiki/Celecoxib
15 E. Holinski-Feder, M. Morak; Hyperplastische Polypen, sessile serratierte Adenome, konventionelle Adenome: Molekulare Pathways und deren klinische Relevanz; J Gastroenterol Hepatol ERKR 2010; 8
16 G.B. Baretton, F. Autschbach, S. Baldus, H. Bläker, G. Faller, H.K. Koch, C. Langner, J. Lüttges, M. Neid, P. Schirmacher, A. Tannapfel, M. Vieth, D.E. Aust; Histopathologische Diagnostik und Differenzialdiagnostik serratierter Polypen im Kolorektum. Ergebnisse einer Konsensuskonferenz der AG „Gastroenterologische Pathologie der DGP“; Pathologe 2011; 32:76–82
17 T Kodama et al., Lymphomatous polyposis of the gastrointestinal tract,
including mantle cell lymphoma, follicular lymphoma and mucosa-associated lymphoid tissue lymphoma; Histopathology 2005, 47, 467–478
Lehrtexte Spezielle Pathologie

Bilder
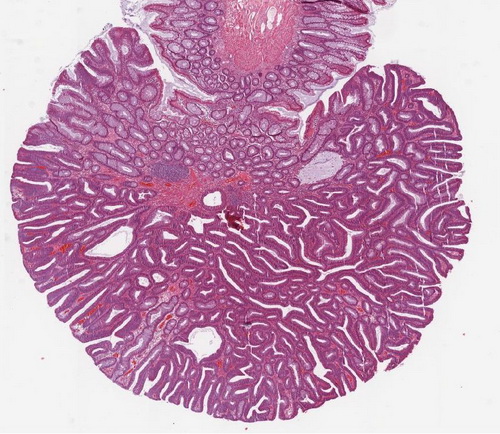
Abb. 507: Adenom bei FAP
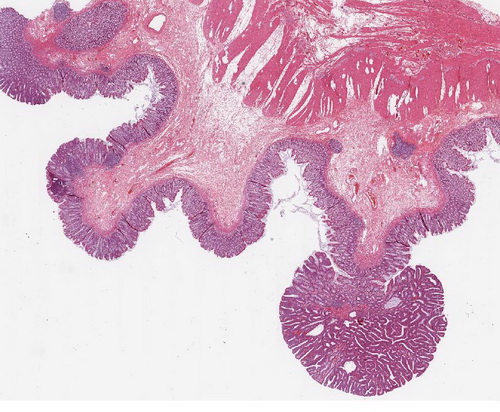
Abb. 558: Erkennbar sind zahlreiche polypöse, adenomatöse Epithelproliferationen, ausgehend von der Kolonmukosa. HE-Färbung.
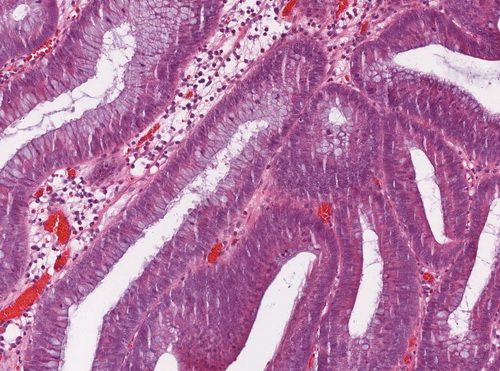
Abb. 559: Auschnitt aus dem Übersichtbild. Erkennbar ist ein mehrreihiges, stäbchenförmiges Oberflächenepithel mit geringen Zellatypien (rechte Bildhälfte)
Kolon und Rektum - weitere Lehrtexte
Kolon und Rektum - Kasuistiken
Organpathologie-Atlas
Weiterführende Literatur
- Kolon und Rektum (34)