16: Medulloblastom (WHO Grad 4)
Morphologie
Makroskopisch sind Medulloblastome heterogen bezüglich ihrer Abgrenzung. Mikroskopisch imponiert vor allem ein sehr zelldichter Tumor mit einer ausgesprochen hohen Kern-Plasma-Relation. Die mitotische Aktivität ist sehr hoch, mikrovaskuläre Proliferationen und Nekrosen sind in der Regel vorhanden. Eine typische Architektur dieser Tumoren ist die neuroblastische Rosette, die aus einer kreisförmigen Anordnung von Tumorzellen um ein virtuelles Lumen besteht.
Kurspräparat
Immunhistologie
Medulloblastome können als Hinweis auf ihren Ausgang von embryonalen Zellen sowohl die neuronalen Marker wie zum Beispiel: Neuronen spezifische Enolase (NSE), Synaptophysin und Neurofilament, als auch den astrozytären Marker GFAP exprimieren.
Genetische und molekulargenetische Befunde
Das Turcot Syndrom (autosomal dominantes Tumorsyndrom mit adenomatösen kolorektalen Polypen und Karzinomen, hervorgerufen entweder durch Keimbahnmutationen in „mismatch-repair“ Genen oder im FAP Gen) und das Gorlin Syndrom (autosomal dominantes Tumorsyndrom mit Basaliomen hervorgerufen durch Keimbahnmutationen im PTCH Gen) sind mit Medulloblastomen assoziiert. Häufigere Mutationen in folgenden Genen wurden gefunden: MYC (Amplifikation) und PTCH.
Neuropathologie

Bilder zum Präparat
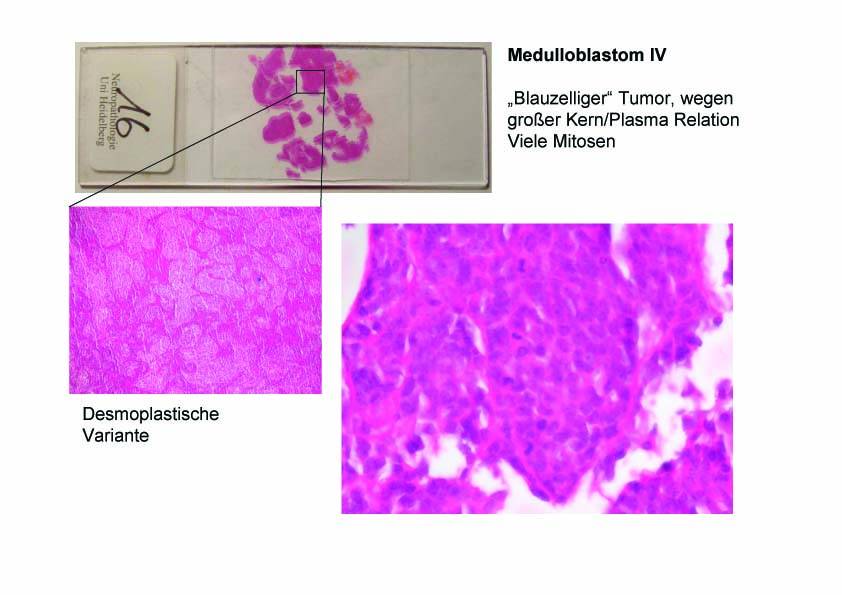
Abb. 55: Medulloblastom (WHO Grad IV): "Blauzelliger" Tumor, wegen großer Kern/Plasma Relation. Viele Mitosen.
Keine weiteren Kurspräparate zu diesem Thema.